
Preparation and characterization of a novel tetrakis(4-hydroxyphenyl)porphyrin–graphene oxide nanocomposite and application in an optical sensor and determination of mercury ions
Rouholah Zare-Dorabei*a,
Rahmatollah Rahimib,
Asgar Koohiab and
Solmaz Zargarib
aResearch Laboratory of Spectrometry & Micro and Nano Extraction, Department of Chemistry, Iran University of Science and Technology, Narmak, Tehran, 16846-13114, Iran. E-mail: Zaredorabei@iust.ac.ir; Fax: +98 21 77491204; Tel: +98 21 77240516
bDepartment of Chemistry, Iran University of Science and Technology, Narmak, Tehran, 16846-13114, Iran
First published on 26th October 2015
Abstract
In this work, for the first time we report a highly specific and sensitive mercury ion (Hg2+) optical sensor (optode). The optode was prepared by recently synthesized nanocomposites, based on 5,10,15,20-tetrakis(4-hydroxyphenyl)porphyrin (THPP) and graphene oxide nanosheets. In this nanocomposite, porphyrin was stabilized on the graphene oxide nanosheets. X-ray diffraction (XRD), UV-vis spectroscopy, field-emission scanning electron microscopy (FE-SEM) and FT-IR were employed to characterize the prepared nanocomposite. In the prepared optical chemical sensor, the formation of a Hg2+–ionophore complex between the mercury ions and the membrane phase, changes the UV-vis absorbance of the sensor. To improve the sensor response, various experimental parameters such as pH, concentration of THPP and graphene oxide nanosheets in the prepared nanocomposite were optimized. This optode exhibited a linear range of 6.0 × 10−9 to 6.0 × 10−5 mol L−1 Hg(II) with a detection limit of 3.2 × 10−9 mol L−1 and a response time of ∼210 s. It manifests advantages of low detection limit, fast response time, wide dynamic range, good reversibility, high reproducibility and also remarkable selectivity regarding the number of transition metals ions (i.e. Pb2+,Cr3+, Zn2+, Cd2+, Cu2+, Fe2+, and Ni2+). This optode was applied for determining Hg(II) ions in water samples.
Introduction
Due to indiscriminate disposal of wastewater, heavy metal pollution is a worldwide environmental concern. Wastewaters from many industries such as metallurgy, chemical manufacturing, and battery manufacturing contain many kinds of toxic heavy metal ions. Mercury is a highly toxic heavy metal which leads to human health problems and environmental contamination.1 The toxicity of mercury is more than lead and arsenic, which leads to neuropsychiatric disorders, infertility in men and women as well as premature aging. Currently, due to increasing utilization of mercury compounds in industry, Hg(II) detection has gained much attention.2–5Design of monitoring systems such as optical chemical sensors for the determination of very low concentrations of mercury ions in water samples can be a very useful and effective approach in the control and removal of pollutants from the environment.6,7
In recent years, extensive research is allocated to optical chemical sensors.8–10 Optical chemical sensors are used in different contexts such as environmental, clinical and industrial analysis. The application of optical chemical sensors is highly regarded especially for the determination of trace amounts of heavy metal ions in environment and a variety of water and food samples.11,12
Optical sensors are a type of chemical sensors which act on the basis of changes in the optical properties. Optical properties are changed due to the interaction between the analyte and the receptor. The main part of the optical sensors is a sensitive membrane which contains the detector. For the design of optical chemical sensor a sensitive layer such as dye indicator (i.e. porphyrin compounds) must be established onto a polymer matrix such as PVC, MIP, and cellulose acetate.2,3
Porphyrins have been studied as probe molecules in different chemical sensors.13 Porphyrins are sensitive to metal ions and act as strong ligands. Electron pairs of nitrogen atoms in the center of the porphyrin ring keep the metal ions and therefore the complex of porphyrin is formed.14–20
The characteristic peaks of porphyrin in the UV-vis spectra is a strong absorption peak (Soret band) in the range of 400–420 nm and four weak absorption peaks (Q bands) in the range of 500–700 nm. These peaks relate to the π → π* electron transfer of porphyrin unsaturated ligand. In metalloporphyrin, relative to porphyrin, the Soret band shifts to higher wavelengths and its intensity is reduced. This behavior represents the sensing properties of porphyrin. Therefore, porphyrins are a good candidate to use as a detector in the optical sensors.
Choosing a proper substrate and detector stabilizer, is a very important issue as another part of the membrane components in the optical sensors. A proper substrate should have suitable functional groups to bind and stabilize the detector on the plastic or glass fulcrum. Therefore, the principle objective of the present research is to use a proper substrate to stabilize the porphyrin detector. Graphene oxide nanosheets with carboxyl, hydroxyl and epoxy functional groups are able to binding and stabilizing the detector on the Fulcrum.21–23 In addition, porphyrin with high coordination ability to metal ions is a suitable detector to determine low concentration of these metal ions.
In this work, a new optical chemical sensor based on porphyrin and graphene oxide nanosheets was designed. The optical sensor was prepared by recently synthesized nanocomposites based on 5,10,15,20-tetrakis(4-hydroxyphenyl)porphyrin (THPP) and graphene oxide nanosheets. It is expected that porphyrin as the strong ligand and with proper optical properties efficiently determines mercury ions. To improve the sensor response, the various experimental parameters such as pH, concentration of the THPP and the graphene oxide nanosheets in the prepared nanocomposite were optimized.
Experimental
Materials
All reagents were of the best available analytical reagent grade and supplied from Merck Co., except HgCl2 which was supplied from Aldrich Co. Buffer solutions (0.10 M) were prepared from phosphoric acid/sodium hydroxide solutions and the pH were adjusted by addition of sodium hydroxide solution. A stock solution of 10−3 M mercury ion solution was prepared by weighing 0.0811 g of HgCl2 and setting the volume to 250 ml. Lower concentrations were prepared by several dilution of the stock solution with phosphate buffer, pH 7.5.Synthesis of graphene oxide–tetrakis(4-hydroxyphenyl)porphyrin (GO–THPP)
 | ||
| Scheme 1 The schematic illustration of the THPP synthesis. | ||
 | ||
| Scheme 2 The schematic illustration of the GO–COCl preparation. | ||
Synthesis of covalently attached porphyrin graphene oxide nanosheets nanocomposite (GO–THPP)
Briefly, in a typical reaction, 3 ml of triethyl amine and 15 ml of DMF were added to a mixture of 30 mg GO–COCl and 60 mg THPP at 80 °C for 72 h under a nitrogen atmosphere. Then, the solution was cooled to room temperature, and then poured into 250 ml diethyl ether to precipitate the product. The precipitate was collected by centrifuging at 8000 rpm for 30 min. The supernatant which contains unreacted THPP, was discarded and the precipitate was washed thoroughly. After adding another 100 ml of diethyl ether, the mixture was sonicated for 5 min and then centrifuged at 8000 rpm for 30 min to collect the GO–THPP. Finally, the precipitate was washed with CHCl3 five times following the above procedure. The schematic illustration of covalently linked GO–porphyrin nanocomposites is shown in Scheme 3.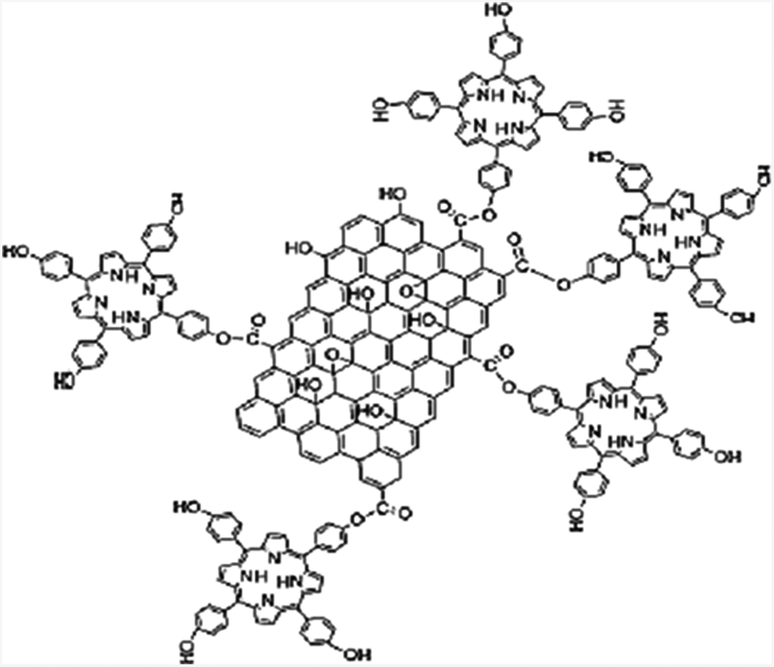 | ||
| Scheme 3 The schematic illustration of the prepared Gr–THPP. | ||
Structural and spectroscopic characterization of the prepared compounds
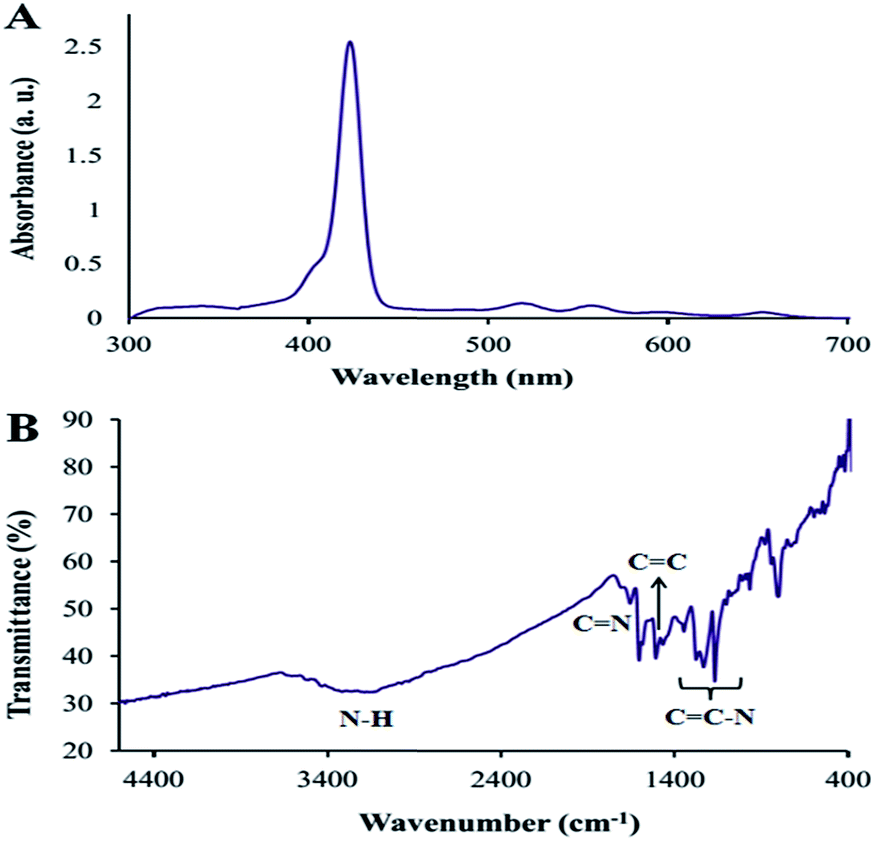 | ||
| Fig. 1 (A) UV/vis absorption spectrum of THPP in DMF and (B) FT-IR spectrum of THPP. | ||
The FT-IR spectrum of this compound showed the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N vibration at 1468 cm−1 and the N–H stretching bond at 3050–3150 cm−1. In addition, the peak corresponding to aromatic ether at 1660 cm−1 and the OH peak at about 3300 cm−1 are shown in Fig. 1B.
N vibration at 1468 cm−1 and the N–H stretching bond at 3050–3150 cm−1. In addition, the peak corresponding to aromatic ether at 1660 cm−1 and the OH peak at about 3300 cm−1 are shown in Fig. 1B.
O group at 1730 cm−1 and CC at 1625 cm−1, which indicates that large amounts of oxygen-containing functional groups exist on graphene oxide nanosheets.
 | ||
| Fig. 2 (A) FT-IR spectrum and (B) XRD pattern of (a) graphite and (b) GO, (C) UV-vis absorption of GO in DMF. | ||
The XRD pattern of graphite and graphene oxide nanosheets are shown in Fig. 2B. For XRD, the interlayer spacing of the materials is proportional to the degree of oxidation. The diffraction peak at 2θ = 26.60° which corresponds to the normal graphite spacing (002) of graphite plane, disappears in the graphene oxide nanosheets. Therefore, no impurity of graphite precursor was remained in the obtained graphene oxide nanosheets. In the XRD pattern of graphene oxide nanosheets, the peak centered at 2θ = 9.2° is assigned to (002) inter-planar spacing of ca. 9.6 Å.
The optical absorption measurements were carried out by UV-vis spectrometry. Fig. 2C shows the optical absorption spectrum of graphene oxide nanosheets. Graphene oxide nanosheets shows an absorption peak at 320 nm originating from the π → π* plasmon of CC which agrees with the literature value.27
The SEM image (Fig. 3) shows that few layered graphene oxides have been formed. Although the SEM image does not estimate the layer numbers of the graphene oxide nanosheets, exactly.
 | ||
| Fig. 3 SEM images of GO nanosheets. | ||
O stretching of the residual COOH groups.28
 | ||
| Fig. 4 (A) FT-IR spectrum and (B) XRD pattern of GO–THPP, (C) UV-vis absorption of GO–THPP. | ||
After covalent functionalization of graphene oxide nanosheets with porphyrins, a new peak at around 1582 cm−1 was appeared corresponding to the CC vibrations of porphyrins. In comparison to the FT-IR spectra of GO–COCl and THPP, in GO–THPP the peak of the CO group appeared at 1168 cm−1 which confirmed that the porphyrin molecules are covalently bonded to the graphene oxide nanosheets via carboxylic acid linkage. In addition, the peak around 1707 cm−1 is assigned to the bending vibration of the CN of the porphyrin ring.
The partial reduction of graphene oxide to graphene in the GO–THPP nanocomposite formation was also verified through XRD analysis. The XRD pattern of GO–THPP shown in Fig. 4B. The broadened peak in the range of 2θ = 17–25°in the GO–THPP pattern is related to the characteristic peak of graphene and graphene oxide. Therefore, in the preparation of GO–THPP some part of graphene oxide was reduced to the graphene.
The optical absorption measurements is a powerful method to determine the presence of porphyrin in nanocomposites.29 Fig. 4C shows UV-vis spectrum of graphene oxide–porphyrin nanocomposites in DMF. In this spectrum the presence of GO peak around 250 nm and the characteristic peaks of porphyrin (Soret and four Q peaks) can be valid evidence for the presence of porphyrin and GO in the GO–THPP nanocomposite.
The scanning electron micrographs (SEM) for the GO–porphyrin nanocomposite materials demonstrate that a homogeneous system with a micrometer order of magnitude was obtained. Fig. 5 shows the selected micrographs for GO–THPP nanocomposite and graphene oxide nanosheets. A clear difference between micrographs of graphene oxide nanosheets and GO–THPP is observed. As shown in Fig. 5B, a three-dimensional network of randomly oriented sheet-like structures are exhibited after the composition of GO with porphyrin.
 | ||
| Fig. 5 SEM images of (A) GO nanosheets and (B) GO–THPP. | ||
Membrane preparation
A mixture of 0.6 mg GO–THPP nanocomposites, was dispersed in 1.0 ml of DMF. The membrane solution was homogenized with a magnetic stirrer for 15 min. A plexiglas slides with 9 mm × 40 mm dimensions were fitted into standard spectrophotometer cells. The slides were cleaned with ethanol, then water and finally dried in an oven at 70 °C. The membranes were cost by pipetting 10 μL of the membrane solution onto a plexiglas slide and spread with a spin coater to deposit a uniform thin film on the glass substrates.Measurement procedure
The membrane was conditioned by inserting it into a cell, including 3 ml of phosphate buffer (pH 7.5). After 5 min, the membrane absorbance was measured at 426 nm. Then, the cell was filled with the Hg(II) standard solution and after 210 s its absorbance was measured in the same wavelength. The membrane response is defined as the ratio of the concentration of the unprotonated form of the chromoionophore [C] to the total amount which is presented in the membrane [Ctot]:
 | (1) |
Then, the value was calculated by the absorbance measurements at the λmax of the protonated form of the chromoionophore as:
 | (2) |
Results and discussion
Principle of the operation
Tetrakis(4-hydroxyphenyl)porphyrin (THPP) contains a nitrogen donor atom which could form internal bonds with soft metal ions such as Hg(II). THPP (Scheme 1) has high affinity to make a complex with Hg(II) ions. Hg(II) ions form a complex with the ligand when the organic membrane contacts with the aqueous solution; therefore, the following ion-exchange reaction takes place:| H2(L)(m) + Hg2+(aq.) ↔ (L)Hg(m) + 2H+(aq.) | (3) |
In this equation, ‘L’ is the ligand. It can be seen that by the addition of the mercury ions in the aqueous solution, the chromoionophore in the organic membrane is more deprotonated. Fig. 6 shows the absorption spectra of the optode with different concentrations of Hg(II) in the range 1.0 × 10−14 to 1.0 × 10−3 mol L−1.
 | ||
| Fig. 6 Absorption spectra of the optical sensor in phosphate buffer solution (pH 7.5) containing different concentrations of Hg(II) as: (1) blank solution (buffer); (2) 1.0 × 10−14; (3) 1.0 × 10−13; (4) 1.0 × 10−12; (5) 1.0 × 10−11; (6) 1.0 × 10−10; (7) 1.0 × 10−9; (8) 1.0 × 10−8; (9) 1.0 × 10−7; (10) 1.0 × 10−6; (11) 1.0 × 10−5; (12) 1.0 × 10−4; (13) 1.0 × 10−3 mol L−1 Hg(II). | ||
This figure indicates that by the addition of Hg(II) to the solution, the absorption of the membrane at 426 nm decreased. The response of the membrane is defined as the ratio of the concentration of the unprotonated form of the ligand [C] to the total amount which is presented in the membrane [Ctot], i.e. α = [C]/[Ctot]. The following equation gives the relative fraction of the deprotonated form of the chromoionophore:
 | (4) |
Effect of sample solution pH
The pH influence on the response of the proposed optical sensor was studied in the range of 3.5–9.5 by changing the universal buffer. As it can be noticed in Fig. 7, as the pH of the solution is increased, the response of the membrane to Hg(II) ions is increased. The sensor response is almost constant in the pH range of 7–8. A maximum value in the optical sensor response was obtained at pH value of 7.5. This pH was selected for the subsequent investigations. | ||
| Fig. 7 The pH effect on the optode film response. | ||
The observed drift at higher pH values could be attributed to the hydrolysis of mercury ions in the solution which decrease the Hg(II) ion concentration in the solution and inhibits the formation of a complex between Hg(II) ions and the THPP. Thus, the optode response decreases. At pH < 6, the heteroatoms of the ligand in the membrane of the optode are protonated and could not form complex with Hg(II) ions in the solution.
Response time
In this research, the optode film was found to reach 95–99% of the final signal at 180–210 s, depending on mercury ion concentration. Fig. 8 shows the time course for the absorption intensity of the membrane at 426 nm. The response time was tested by recording the absorbance change from a pure buffer (pH = 7.5) to a buffered Hg(II) solution of 1.0 × 10−5 M. | ||
| Fig. 8 Response time curve of the optode at 426. | ||
The response time of the optical sensor depended on the organic membrane thickness, the organic membrane composition, the concentration of the measuring ion and the pH of the measurement. The response time of the optode in a higher concentration of Hg(II) was restricted by the time requested for the analyte diffusion from the bulk of the solution toward the membrane. For highly diluted solutions, the limiting step is the convective mass transport from the bulk of the aqueous solution to the membrane.
Dynamic range
Under the optimum conditions, calibration graphs for Hg(II) was constructed by the response function (1−α) values were obtained at 426 nm in different Hg(II) concentrations. Fig. 9 shows the calibration graph revealed that the signal is linear in the range of 6.0 × 10−5 to 6.0 × 10−9 with a regression equation of (1−α) = −0.1211(log[Hg(II)]) − 0.2106, (R2 = 0.9976). The detection limit (3Sb/m) of the method was calculated as 3.2 × 10−9, which indicates the proposed sensor is more efficient and sensitive for mercury ion.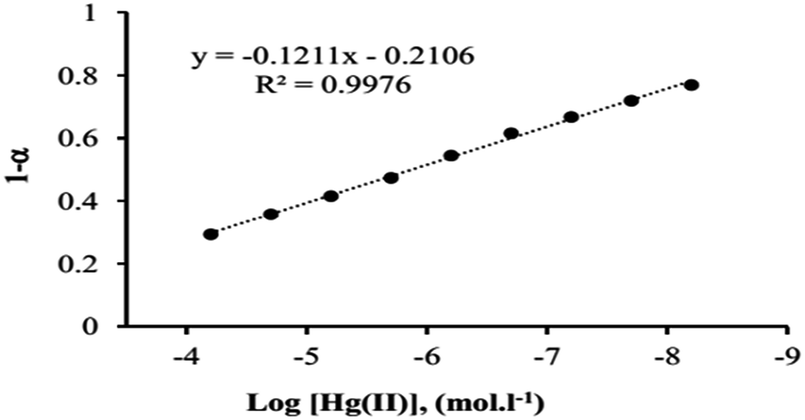 | ||
| Fig. 9 Calibration curve for determination of Hg2+ in the optimum conditions. | ||
Optode regeneration (reversibility)
For suitable performance of the optode membrane, its absorbance change must be reversible. Primary tests were conducted with a number of reagents to reverse the absorbance of Hg(II) complex. Complexing agents such as EDTA and sulfosalicylic acid have a partial reverse effect, and prolonged exposure to them have no further improvement in reversibility of the optode. EDTA (0.2 M) was concluded to be the best reagent, giving a short regeneration time (less than 3 min). To evaluate the discrepancies in the response for successive runs using a single optode membrane, the reproducibility was evaluated by performing 5 determinations with the same Hg(II) standard solution. For 1.0 × 10−5 M Hg(II) a drift of about 4.07% was obtained in the response of the optode. Fig. 10 shows the absorbance changes versus time for the optode membrane.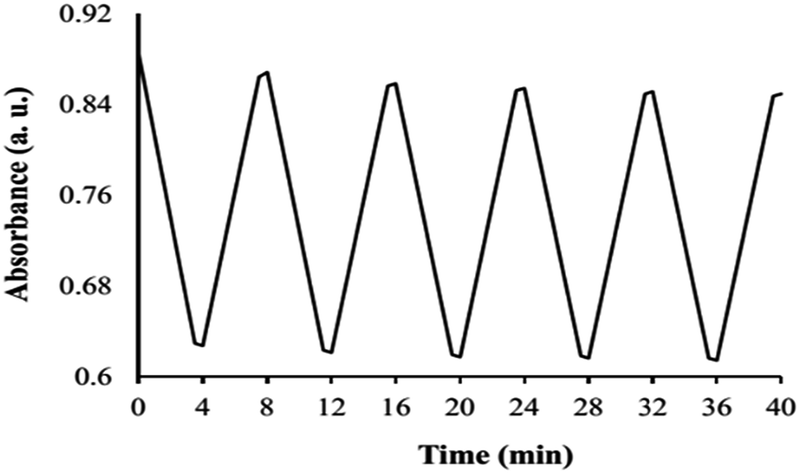 | ||
| Fig. 10 An absorbance variation of the membrane at 426 nm for repeatedly exposing into 1.0 × 10−5 M Hg(II) solution and 0.2 M EDTA solution. | ||
Lifetime and stability
The lifetime of the optode was determined by adding a buffer solution (pH = 7.5) into the cuvette, containing the membrane. The signal was recorded at a wavelength of 426 nm over a period of time (about 10 h). No significant indicator loss occurred during that time. When the film was exposed to light, no drift in the signal took place and the optode was found to be stable during the experiment with no indicator leakage. Stability of film response was investigated over six weeks under ambient conditions, which is indicated that the membrane was stable over this period.Effect of the foreign ions
The interference for a number of common species of the absorbance determination of Hg(II) ions was investigated using the prepared sensor. To determine the selectivity of the optode membrane, the optode membrane was tested under mercury ions concentration of 3 × 10−3 M in the presence of other metal ions. Limit of tolerance was taken as the concentration causing an error of ±5% in the mercury ions assay. The results are summarized in Table 1. The surprisingly high selectivity of the optode membrane for Hg(II) ions over other cations used, most probably arises from the strong tendency of the THPP for formation of stable complexes with Hg(II) ions.| Species | Relative error% | Species | Relative error% |
|---|---|---|---|
| Cd2+ | 4.6 | Na+ | 1.06 |
| Pb2+ | 3.7 | Fe2+ | 2.11 |
| Ni2+ | 3.1 | Cr3+ | 2.12 |
| Co2+ | 0.34 | Ti+ | 0.5 |
| Cu2+ | 1.06 | Cs+ | 0.8 |
| Zn2+ | 1.76 | Ag+ | 3.2 |
| K+ | 1.009 | Mn2+ | 2.9 |
| Mg2+ | 2.3 | Ca2+ | 3.4 |
| NH4+ | 1.1 | Al3+ | 3.9 |
Repeatability and recovery tests
The repeatability was examined by preparing 15 different membranes from the same mixture and measuring the absorbance of each membrane at 426 nm in 6.0 × 10−6, 6.0 × 10−7 and 6.0 × 10−8 mol L−1 Hg(II) (five replicate determinations) in the buffer solutions at pH 7.5. The RSD% for 6.0 × 10−6, 6.0 × 10−7 and 6.0 × 10−8 mol L−1 were 4.33%, 3.34% and 5.09%, respectively (Table 2). This result shows that the sensor has a good repeatability.| Standard concentrations of mercury (M) | Average absorption (n = 5) | Average Hg(II) found with optode (M) (n = 5) | RSD (%) (n = 5) | Recovery (%) |
|---|---|---|---|---|
| 6.0 × 10−6 | 0.7 | 6.28 × 10−6 | 4.33 | 104 |
| 6.0 × 10−7 | 0.7384 | 5.96 × 10−7 | 3.34 | 99 |
| 6.0 × 10−8 | 0.7758 | 6.017 × 10−8 | 5.09 | 101 |
| 6.00 × 10−8 (Tehran river water) | 0.769 | 5.89 × 10−8 | 4.91 | 98 |
The recovery tests were performed using these three different concentrations. The test for each sample was carried out in triplicate measurements and the results are given in Table 2. As it is evident from Table 2, Hg(II) recovery values were between 98 and 104.
Conclusions
In this work, for the first time a highly specific and sensitive optical sensor (optode) was developed based on the tetrakis(4-hydroxyphenyl)porphyrin (THPP) stabilized with graphene oxide nanosheets for the determination of ultra-trace amounts of Hg(II) ions. The sensor was easily prepared, readily regenerated with a EDTA solution and demonstrated a long lifetime. The optode response was concluded to be reproducible with a good Hg(II) selectivity over other potentially interfering ions. Since the optical sensor required no solvent extraction, it could compete satisfactory with the standard optical fibers. The characteristics of the proposed Hg(II) ions optical chemical sensor were compared with other determination methods of literature in Table 3.30–32 This optode was applied to the determination of Hg(II) in different concentrations in water samples with good precision and accuracy.| Method | DLa (M) | LRb (M) | RSD% | Samples | Ref. |
|---|---|---|---|---|---|
| a Detection limit.b Linear range. | |||||
| SPE-AAS | 3.0 × 10−13 | 6.0 × 10−13 to 4.0 × 10−13 | 4.7 | Water | 30 |
| Colorimetric sensor | 4.0 × 10−9 | 0 to 333.0 × 10−9 | — | Water | 31 |
| Colorimetric sensors | 4.0 × 10−8 | 2.5 × 10−6 to 3.5 × 10−5 | 3.24–4.53 | Water | 32 |
| Optical sensor | 3.2 × 10−9 | 6.0 × 10−5 to 6.0 × 10−9 | 4.41 | Water | This work |
Acknowledgements
The financial support of this study by Iran University of Science and Technology and Nano Technology Initiative Council are gratefully acknowledged. The author acknowledges financial support from the Iran National Science Foundation (INSF).References
- Z. Li, S. Xia, J. Wang, C. Bian and J. Tong, J. Hazard. Mater., 2016, 301, 206–213 CrossRef CAS PubMed.
- A. R. Firooz, M. Movahedi and A. A. Ensafi, Sens. Actuators, B, 2012, 171–172, 492–498 CrossRef CAS.
- Z. Yan, M. F. Yuen, L. Hu, P. Sun and C. S. Lee, RSC Adv., 2014, 4, 48373–48388 RSC.
- K. Alizadeh, R. Parooi, P. Hashemi, B. Rezaei and M. R. Ganjali, J. Hazard. Mater., 2011, 186, 1794–1800 CrossRef CAS PubMed.
- V. S. Basha, S. V. babu, G. Narasimha and K. H. Reddy, Anal. Chem.: Indian J., 2013, 13, 182–186 CAS.
- A. R. Firooz, A. A. Ensafi, K. Karimi and R. Khalifeh, Sens. Actuators, B, 2013, 185, 84–90 CrossRef CAS.
- H. Tavallali, E. Shaabanpur and P. Vahdati, Spectrochim. Acta, Part A, 2012, 89, 216–221 CrossRef CAS.
- K. S. C. Kuang, Sens. Actuators, A, 2015, 229, 59–67 CrossRef CAS.
- R. Zare-Dorabei, K. Dashtian and V. Jalalat, IEEE Sens. J., 2015, 15, 6715–6721 CrossRef.
- M. Saleem and K. H. Lee, RSC Adv., 2015, 5, 72150–72287 RSC.
- J. Janata and A. Bezegh, Anal. Chem., 1988, 60, 62R–74R CrossRef CAS PubMed.
- M. R. Ganjali, R. Zare-Dorabei and P. Norouzi, Sens. Actuators, B, 2009, 143, 233–238 CrossRef.
- V. K. Gupta, A. K. Jain, Z. Ishtaiwi, H. Lang and G. Maheshwari, Talanta, 2007, 73, 803–811 CrossRef CAS PubMed.
- R. Yang, K. Li, K. Wang, F. Liu, N. Li and F. Zhao, Anal. Chim. Acta, 2002, 469, 285–293 CrossRef CAS.
- W. H. Chana, R. H. Yang and K. M. Wang, Anal. Chim. Acta, 2001, 444, 261–269 CrossRef.
- P. Kumar and Y. B. Shim, J. Electroanal. Chem., 2011, 661, 25–30 CrossRef CAS.
- I. Leray, M. C. Vernieres, R. L. Saibi, C. B. Charreton and J. Faure, Sens. Actuators, B, 1996, 37, 67–74 CrossRef CAS.
- H. K. Lee, K. Song, H. R. Seo, Y. K. Choi and S. Jeon, Sens. Actuators, B, 2004, 99, 323–329 CrossRef CAS.
- K. Kilian and K. Pyrzynska, Talanta, 2003, 60, 669–678 CrossRef CAS PubMed.
- R. Rahimi, R. Zare-Dorabei, A. Koohi and S. Zargari, Proceedings of the 18th International Electronic Conference on Synthetic Organic Chemistry, Lugo, 2014 Search PubMed.
- X. Gong, Y. Bi, Y. Zhao, G. Liu and W. Y. Teoh, RSC Adv., 2014, 4, 24653–24657 RSC.
- Q. Xiang, J. Yu and M. Jaroniec, Chem. Soc. Rev., 2012, 41, 782–796 RSC.
- V. Georgakilas, M. Otyepka, A. B. Bourlinos, V. Chandra, N. Kim, K. C. Kemp, P. Hobza, R. Zboril and K. S. Kim, Chem. Rev., 2012, 112, 6156–6214 CrossRef CAS.
- S. Shanmugathasan, C. Edwards and R. W. Boyle, Tetrahedron, 2000, 56, 1025–1046 CrossRef CAS.
- M. B. M. Krishna, N. Venkatramaiah, R. Venkatesan and D. N. Rao, J. Mater. Chem., 2012, 22, 3059–3068 RSC.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- M. A. Khaderbad, V. Tjoa, T. Z. Oo, J. Wei, M. Sheri, R. Mangalampalli, V. R. Rao, S. G. Mhaisalkar and N. Mathews, RSC Adv., 2012, 2, 4120–4124 RSC.
- H. Zhang, X. Lv, Y. Li, Y. Wang and J. Li, ACS Nano, 2010, 4, 380–386 CrossRef CAS PubMed.
- R. Rahimi, M. Mahjoub Moghaddas and S. Zargari, J. Sol-Gel Sci. Technol., 2013, 65, 420–429 CrossRef CAS.
- E. Ziaei, A. Mehdinia and A. Jabbari, Anal. Chim. Acta, 2014, 850, 49–56 CrossRef CAS PubMed.
- J. Duan, H. Yin, R. Wei and W. Wang, Biosens. Bioelectron., 2014, 57, 139–142 CrossRef CAS PubMed.
- P. Jarujamrus, M. Amatatongchai, A. Thima, T. Khongrangdee and C. Mongkontong, Spectrochim. Acta, Part A, 2015, 142, 86–93 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |