
A simple technique for performing evaporation of quaterthiophene below the melting temperature for vapour phase polymerisation and physical vapour deposition†
David Mayevsky*a,
Jacob Tosadob,
Christopher D. Eastonc,
Chun Hin Nga,
Michael S. Fuhrerb and
Bjorn Winther-Jensena
aDepartment of Materials Engineering, Monash University, Clayton, VIC 3800, Australia. E-mail: david.mayevsky@monash.edu
bDepartment of Physics, Monash University, Clayton Campus, VIC 3800, Australia
cCSIRO Manufacturing Flagship, Clayton, VIC 3168, Australia
First published on 16th November 2015
Abstract
By adjusting the molecular ordering of the evaporant used for vapour deposition, anappreciable evaporant partial pressure was achieved at temperatures far below the evaporant melting temperature with evaporation occurring over 100° below the melting temperature. The molecular ordering was adjusted by dissolving or dispersing the evaporant (MW > 300) in an ionic liquid or a high MW non-vaporisable polymer (MW ∼ 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000).
000).
The field of organic electronics has significantly grown as a consequence of creating versatile processing techniques to match the requirements of new device designs. The most common strategy to enable processing is by engineering solubility through chemical modification polymers. Alternative processing methods are also prevalent, which include chemical vapour deposition of small organic molecules, instead of polymers, and polymerisation (via chemical or electrochemical means) of the monomers at the location of use. Developing new processing techniques facilitates device manufacturing using new materials, or materials with better morphological properties1,2 which in turn is can lead to engineering devices with improved performance.
In particular organic layers manufactured by evaporative techniques have can exhibit increased performance,3 and this is asserted to be a consequence of better molecular ordering and structural regularity1,2,4 However these techniques require having the capacity to evaporate the organic material, and commonly, larger molecular weight organics decompose instead of evaporating. In contemporary research is has become increasingly popular to use larger organics.4 This inability to evaporate some materials limiting factor is a limiting for the use of evaporative techniques. This work addresses the issue of being unable to evaporate larger organic molecules such that evaporative techniques can be used for new materials.
Vapour phase polymerisation (VPP) and physical vapour deposition (PVD) are commonly used techniques for manufacturing of homogenous organic semiconductor thin films.5 VPP requires appreciable partial pressure of a polymerisable monomer in an enclosed chamber. In the case of PVD by sublimation; the temperature must be high enough and the pressure low enough for a sublimation to occur. The most commonly used method, to achieve either evaporation or sublimation, is by applying a temperature or reducing the surrounding pressure of the material that will be deposited. The ability to deposit higher molecular weight organics uniformly is a challenge for PVD processing community.6
Evaporation can be modelled by the Clausius–Clapyron7 relation (eqn (1)). Where ΔP is the pressure difference as a consequence of changing the ΔT. The relation also states that the pressure difference is also related to the ΔS, the entropy of the substance which is evaporating.
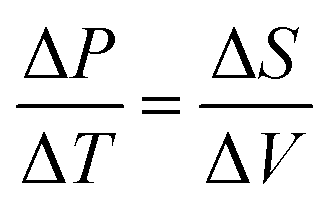 | (1) |
A new technique, which to our best knowledge has not been outlined before, is reported in this communication; where the molecular ordering of a precursor is modified before commencing the evaporation. By adjusting the bonding associated with packing in a crystal structure one can induce positive ΔS by increasing the disorder of the structure, and possibly induce a –ΔH (enthalpy) if there is energy stored in keeping the crystalline structure; which may be a free energy barrier to evaporation.
In other words, this new and simple approach is the technique of highly zeotropic distillation of an evaporant which can be performed by modifying the molecular ordering by solubilising/dispersing the evaporant. Utilising a non-evaporating solvent such as an ionic liquid (IL), which are asserted to have low vapour pressures,8 or a very high molecular weight liquid polymer as a dissolution/dispersion medium. The underlying concept is that by blending this IL or high molecular weight polymer with the evaporant, one can adjust the packing of the evaporant in the blend, therefore reducing the temperature required for the evaporation. It is expected that by using the non-evaporating dissolution medium only the desired monomer will evaporate.
In blending the evaporant with an IL or high molecular weight polymer, for the same temperature, it is expected that the evaporant that is blended will be subject to a +ΔS, compared to the pure unblended state of the same material. The ΔP and ΔS will only be changed for the evaporant, and not for the whole system (ΔP and ΔS are compared for evaporant on its own and for evaporant in non-evaporating liquid). This assumption can be justified because it can be assumed that there is no competing vapour pressure from the high molecular weight liquid. A +ΔS is achieved for the evaporant by blending and consequentially an increase in vapour pressure can be achieved, in accordance to eqn (1).
The same strategy can be used to slow down deposition processes under some circumstances and in some circumstances achieve extremely low deposition rates. In the case of VPP this can be done when attempting to evaporate from the blend at above the monomer melting temperature, where otherwise the deposition rate would be high. This strategy can potentially be useful for achieving controlled partial pressure of multiple evaporants; and consequentially a multiple component deposition can be achieved. This technique could prove particularly useful if the melting temperatures (and thereby the vapour pressures) of the evaporants do not match, and a dual component deposition/polymerisation is required. The ability to match evaporation rates could allow for the evaporative manufacturing of copolymers, which show new electrical properties.9
Herein we report an experimental test case of the sub-melting temperature evaporation of quaterthiophene (QTh) and synthesis of a deposited polyquaterthiophene (PQTh) thin film, where the evaporation and deposition of the film was performed without the aid of a vacuum at 100 °C (116 °C below the 216 °C melting point) using phosphonium 1,2,2,4 tosylate (P1224Tos) or a polyethylene glycol/polypropylene glycol copolymer as a dissolution/dispersion medium. We also report vacuum physical vapour deposition of QTh where the sublimation temperature for the QTh + PEG blended system is well above the sublimation temperature for the evaporant and the deposition process allows for extremely low, controllable deposition rates.
Experimental
Materials
QTh (2,2′:5′,2′′:5′′:2′′′-Quaterthiophene) was purchased from Sigma Aldrich, and used as purchased. PEG20K (MW 20000) was purchased from Fluka [CAS no: 25322-68-3, catalogue number: 81300]. PEG copolymer [referred to in text as PEGC] (poly(propylene glycol)-block-poly(ethlene glycol)-block-poly(propylene glycol)) [CAS no: 9003-11-6, catalogue number: 435481] (Mn ∼ 2700) was purchased from Aldrich. The IL used, P1224TOS, was manufactured by Cytec. Fe(III) PTS (iron(III) paratoluene sulfonate) 40% in butanol was purchased from Yacoo Chemical Reagent.
Vapour phase polymerisation (VPP) method
For the thin film production via VPP, 15 mg QTh was dispersed in either 0.25 ml IL (P1224Tos) or in 0.25 ml of PEGC then 1 ml of tetrahydrofuran (THF) was added to aid dispersion. This THF was boiled off (at 90 °C) after the QTh was well dispersed (before the polymerisation step). A 20 ml vial containing the remaining QTh in IL or QTh in PEGC mixtures was placed in a sealed polymerisation chamber. The substrates used for VPP deposition are coated with an oxidant, for the purpose of performing oxidative vapour phase polymerisation. The Fe(III)PTS 40% oxidant in butanol which was spin coated onto the substrates at 1500 RPM and the excess butanol is evaporated at 70 °C. These substrates are placed in the polymerisation chamber, and evaporation was performed at 100 °C for 50 hours. Samples were washed in ethanol for 24 hours to remove the remaining oxidant. As baseline experiments to ensure only QTh was deposited, the IL and the PEGC were used to perform the VPP experiment without addition of the monomer to this precursor.Physical vapour deposition method
All reported depositions were performed under vacuum at 10−6 Torr, after an overnight degassing. For film formation via PVD, 15 mg QTh was dispersed in PEG20K (MW 20000) in the same method outlined in the VPP section above. PEG20K is solid at room temperature and consequentially takes up less moisture (than the PEGC used previously) from atmosphere; excess moisture would likely evaporate before the QTh evaporant.
For PVD rate measurements; optical/electrochemical characterisation, as well as the temperature calibration of the instrument, the depositions were performed by increasing current through the heating element stepwise, holding for 15 minutes at each temperature point, and recording total deposition quantity every 5 minutes using a built-in quartz crystal microbalance (QCM). In the case of QTh an overnight degassing was performed at 140 °C prior to the deposition due to the inherent fluffiness of the purchased monomer.10 Depositions were performed using pure PEG20K or pure QTh as a comparison, and then using the blend for PEG20K and QTh. These depositions were performed by keeping PEG20K below its apparent boiling point which is 180 °C. Using this method, the same 3 depositions were made, one from pure QTh, one using pure PEG20K and one using QTh + PEG20K.
The temperature of the chamber was calibrated using a thermocouple placed in pure PEG20K, using the same time intervals as used for the depositions. Overnight degassing was performed for all of these samples for process consistency. Depositions were performed onto gold mylar and quartz substrates.
PVD rate measurements for a pure PEG20K sample was prepared by dissolving the PEG20K in THF and then boiling off the THF, as was done with the previous samples. The PVD experiment was performed using identical parameters to the one used for QTh and the mixture of QTh + PEG20K.
Characterisation
Absorption spectroscopy was performed using a Jasco V-670 Spectrophotometer on the films deposited on quartz. Cyclic voltammetry (Princeton Applied Research VMP2Z potentiostat) in a 3 electrode setup was performed in dry propylene carbonate electrolyte with 0.1 M tetrabutylammonium hexafluorphosphate (TBAPF6) supporting electrolyte, in the same method used in previous work.11 This experiment was performed in a nitrogen glove box. Experiments were performed at 25 mV s−1 using a 0.01 M Ag/AgClO4 reference electrode in 0.1 M TBAPF6. Only the first and the 5th cycle of each experiment were reported.Fourier transform infrared spectroscopy was performed using Perkin-Elmer Spectrum100. The polymer films that were manufactured on Quartz for absorbance spectroscopy were soaked in ethanol and scraped off into a vial, remaining ethanol was evaporated at 50 °C, and the samples were placed on the ATR crystal in a powdered form. The IL and the PEGC were simply dropped in their liquid form onto the crystal and measured. Measurements were made using 16 accumulations.
XPS was used to determine the composition of the deposited films. The substrate used for XPS experiments was Goretex sputtered with gold. X-ray photoelectron spectroscopy (XPS) analysis was performed using an AXIS Ultra DLD spectrometer (Kratos Analytical Inc., Manchester, UK) with a monochromated Al Kα source at a power of 72 W (6 kV × 12 mA) for survey spectra and 144 W (12 kV × 12 mA) for high resolution spectra, a hemispherical analyser operating in the fixed analyser transmission mode and the standard aperture (analysis area: 0.3 mm × 0.7 mm) The total pressure in the main vacuum chamber during analysis was typically between 10−9 and 10−8 mbar. Survey spectra were acquired at a pass energy of 160 eV. To obtain more detailed information about chemical structure, oxidation states etc., high resolution spectra were recorded from individual peaks at 40 eV pass energy (yielding a typical peak width for polymers of 0.9–1.0 eV). Each specimen was analysed at an emission angle of 0° as measured from the surface normal. Assuming typical values for the electron attenuation length of relevant photoelectrons the XPS analysis depth (from which 95% of the detected signal originates) ranges between 5 and 10 nm for a flat surface. Since the actual emission angle is ill-defined in the case of rough samples (ranging from 0° to 90°) the sampling depth may range from 0 nm to approx. 10 nm.
Data processing was performed using CasaXPS processing software version 2.3.15 (Casa Software Ltd., Teignmouth, UK). All elements present were identified from survey spectra. The atomic concentrations of the detected elements were calculated using integral peak intensities and the sensitivity factors supplied by the manufacturer.
The accuracy associated with quantitative XPS is ca. 10–15%. Precision (i.e. reproducibility) depends on the signal/noise ratio but is usually much better than 5%. The latter is relevant when comparing similar samples.
Results and discussion
It should be noted that it is apparent that the polymerisation by VPP did not completely occur when using a QTh + PEGC precursor however polymerisation did occur when using the QTh + IL. Henceforth all samples for which polymerisation did not occur in the presence of oxidant will be referred to as vapour deposited (VD).Fig. 1 below shows the UV-Vis absorbance of PVD QTh, PVD QTh from the mixture of QTh + PEG20K, VPP QTh from the QTh + IL mixture and a baseline of a VPP experiment where there was no QTh in the IL (IL only). An optical absorbance from polymerised evaporated monomer can be seen in the VPP PQTh sample from IL mixture at 200 nm and 230 nm. No absorbance was seen in the sample where the same experiment was performed without QTh but using the IL only for evaporation. This indicates that it is indeed QTh evaporating and then depositing for the PVD QTh, PVD QTh from QTh + PEG20K, VPP QTh from the QTh + IL mixture samples. It was found for the QTh + PEG precursors, the PEG was evaporating and depositing on the oxidant. This depletes some of the oxidant and consequentially prevents polymerisation, however it does not stop the evaporation and consequential deposition of QTh.
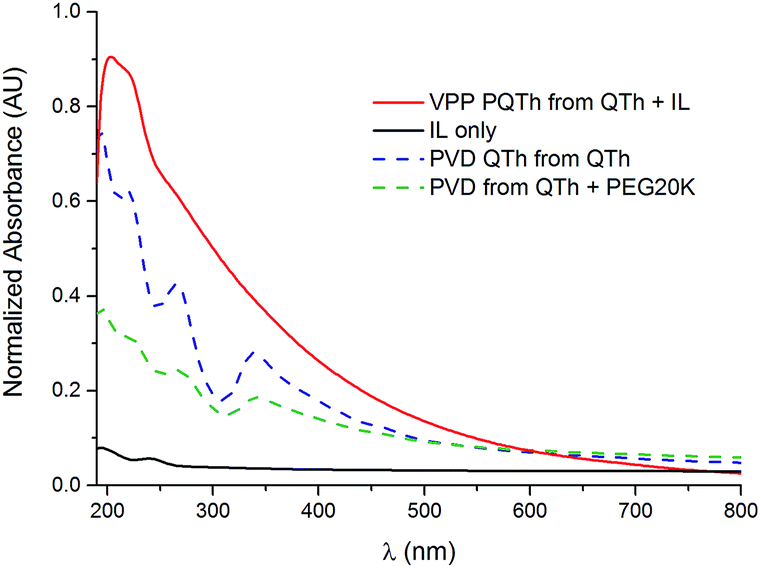 | ||
| Fig. 1 The optical absorbance of PQTh VPPed from the IL (solid red line), baseline of just the deposition from pure IL (solid black line), PVD QTh (blue dashed line) and PVD QTh from QTh + PEG20K (green dashed line). | ||
The deposited oligothiophene from the QTh + IL mixture exhibits a π–π* absorbance peak at wavelengths well below that of polythiophene. This sample did not exhibit the typical peak occurring at ∼490 nm for polythiophene, the observed peak at 200 nm is below literature reported values for absorbance by QTh (390 nm),11 however this can be attributed to the fact that those measurements were made in solution, and not the solid state. The optical absorbance of a drop cast QTh (Fig. S1†) shows peaks occurring at both ∼275 nm and ∼340 nm (as also seen in the PVD samples).
The presence of the oxidant apparently acts as a templating location for the evaporating PEG20C and QTh monomer; as an evidence that this deposition is selectively occurring, deposition occurred only on the oxidant coated side of the glass slide in the chamber when depositing from QTh + PEGC. In Fig. S2† the optical absorbance of PEGC (without any QTh) is shown, it can be seen that PEGC is depositing on the oxidant and presumably coordinating to the Fe(III), and potentially behaving as the templating location for QTh, it is nevertheless clear that QTh did deposit based on the peak at 200 nm.
Based on the optical absorbance experiments it can be seen that in the case of deposition from both of the QTh + IL and QTh + PEG20K mixtures, evaporation of the QTh did occur; and in the case of the IL a pure evaporation and deposition of QTh occurred.
The samples from VPP, VD and PVD were tested electrochemically and as can be seen in Fig. 2a and b. The N doping peaks at ∼−2.6 eV generically correspond to oligothiophenes; as this peak does not shift appreciably with change in chain length.7 This indicates that for all VPP, VD and PVD samples, an oligothiophene was deposited.
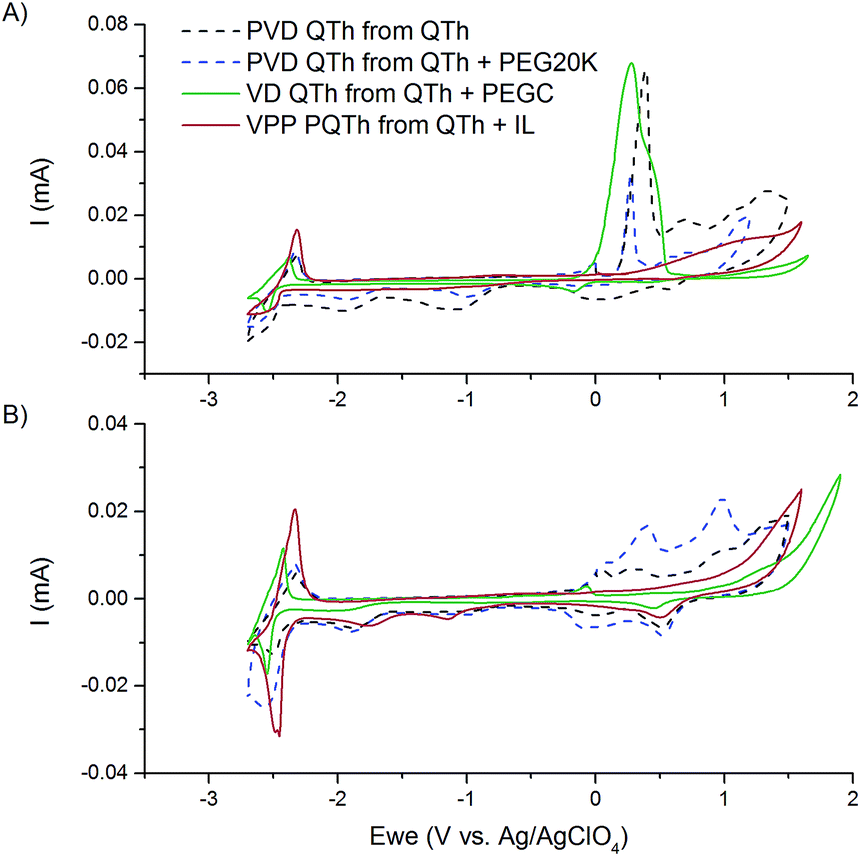 | ||
| Fig. 2 (a) The first cycle and (b) the fifth cycle of the cyclic voltammograms of VPP PQTh from the QTh + IL precursor (solid red curve), VD QTh from QTh + PEGC precursor (solid green curve), PVD QTh from QTh (dashed black curve) and PVD QTh from QTh + PEG20K precursor (dashed green curve). | ||
All films except the one deposited from the IL commence oxidative electropolymerisation at ∼0.2 V vs. Ag/AgClO4 in the first cycle. Meerholz and Heinze12 report the same system commencing electropolymerisation at a slightly lower oxidation potential (0.8 V vs. Ag/AgCl (0.0 V vs. Ag/AgClO4) which can be attributed to the differences in scan rate and the solvent). This oxidation peak exists for the PVD as well as VD samples, indicating that quaterthiophene was deposited and remained unpolymerised in the deposition under all circumstances except in the case of the QTh + IL mixture. In the case of the deposition from the QTh + IL mixture, no oxidation peak corresponding to the polymerisation occurred in the first cycle, indicating that VPP was successfully performed and the QTh was polymerised.
Oxidative peaks are observed at potentials above the oxidative polymerisation potential, and a discrepancy is noticeable between the PVD samples and the VD sample. For the VD sample, these peaks are smaller compared to the oxidative peak and the HOMO/LUMO. It has been asserted that these peaks correspond to multiple electron uptake by the material,12 however, this character would not be different between material that was deposited by PVD and material deposited by VD. Therefore we offer the explanation that this discrepancy is due to the formation of a larger more homogenous crystal structure in the VD sample; where the deposition method is less vigorous than the ballistic deposition of the PVD samples. This would allow for better packing of the QTh, and consequentially require less ‘over potential’ for oxidative polymerisation. This explanation is further supported by the fact that the oxidative peak of the QTh for the first cycle is largest in the VD sample. Upon repeated cycling, at the 5th cycle, the oxidative peak at ∼0.1 eV vs. Ag/AgClO4 disappears (Fig. 2b) indicating that electropolymerisation has occurred.
FTIR was performed in order to characterise the purity of the film manufactured by the VPP process. FTIR was chosen as the method of purity characterisation over a chromatography technique because polythiophene, like many conducting polymers, is insoluble when polymerised, unless there are alkyl chains on the functional groups; this restricts characterisation to solid-state techniques.
Fig. 3A shows that when using both the QTh + IL precursor and the QTh + PEGC precursor, the depositions have very similar vibrational signatures as expected from conjugated thiophenes, with two peaks sitting at ∼1425 cm−1,13 indicating that polythiophene was indeed manufactured. Fig. 3B shows the polythiophene deposited from the QTh + IL precursor, the peak at ∼900 cm−1 is conspicuously absent in the film, however present in the ionic liquid, and we attribute this to P–CH3. The peaks at ∼1150 cm−1 and 1200 cm−1 are assigned to the tosylate from the oxidant that remains as a dopant in the PQTh film and not the tosylate from the ionic liquid as explained below. Fig. 3C shows the PQTh formed from the PEGC precursor and the pure PEGC. It can be seen that these two samples have completely different vibration signatures, in particular, no peak at 1090 cm−1, corresponding to C–O stretching, suggesting that no PEG copolymer remains in the film.
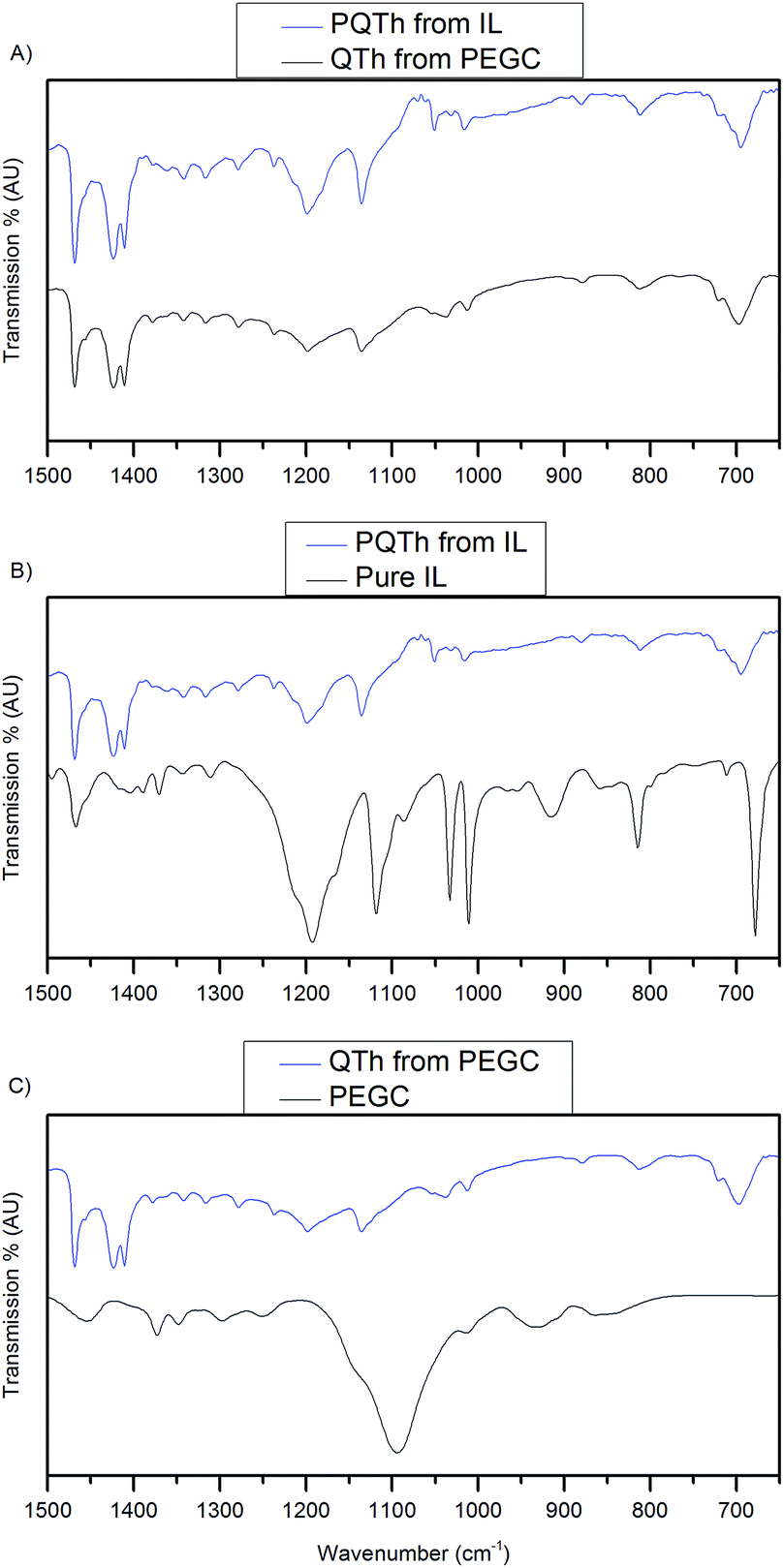 | ||
| Fig. 3 Comparison of FTIR spectra between (a) PQTh from the IL and QTh from the PEG copolymer, (b) PQTh from the IL and the IL and (c) QTh from the PEGC and pure PEGC. | ||
We suggest that tosylate remains in the film as dopant, from the oxidant precursor, and the vibrational signatures that are common between the film and the precursor are due to this tosylate. Supporting this notion, the PQTh manufactured from the IL and the PEG copolymer both have indistinguishable vibration spectra, indicating that this tosylate is present even when the IL is not used as a component of the precursor.
Based on the optical absorbance, vibrational spectroscopy and electrochemical experiments, it can be concluded that for the VPP experiment from the IL oxidative polymerisation was successfully performed on the evaporating QTh, at 116 °C below the melting temperature of QTh. In the case of the QTh + PEGC, evaporation of QTh occurred, but incomplete polymerisation occurred due to evaporating and depositing PEG interfering with the Fe(III) salt which is functioning as an oxidant. Nevertheless the cyclic voltammograms of the VD QTh from QTh + PEGC, the PVD QTh and PVD QTh from QTh + PEG20K all exhibited the same electropolymerisation behaviour as well as the same N doping peak indicating that, under all circumstances, QTh was evaporated and only in the case of the IL mixture did it successfully polymerise.
XPS was used to analyse the relative amounts of sulphur and carbon deposited in the QTh and QTh PEG20K samples prepared by PVD on gold coated Goretex. The high resolution scan over the S 2p region (Fig. S3†) shows that only one oxidation state of sulphur is present, indicating that the thiophene structure remains unbroken in deposition. In order to verify the purity of the deposition from PEG (Table S1†) we assume a fixed C/F ratio for all of the substrates in all 3 samples. C which is contained in the substrate was subtracted from the total C; and thereafter the C:S ratio of the deposited film was calculated. It is found that the C:S ratio for the QTh sample is 4.9 and the QTh PEG sample is 5.0, indicating that the purity of that which is deposited in both circumstances is uniform although a ratio of 4 was to be expected from the structure of QTh.
An exploration was undertaken to measure the deposition rate for PVD to determine the effect of blending with PEG20K. The deposition rate in Fig. 4A shows that the deposition of QTh from the mixture of QTh + PEG20K was commencing at 140 °C, which is the same temperature as reported by Kloc et al.10 Furthermore, evaporation is significantly slower for the mixture of QTh + PEG20K than the PVD of QTh alone. The method allowed for a significantly more controllable deposition with controllable rates of <5 Å min−1 over a range of temperatures. This same level of control was not achievable with QTh only which we suspect that it is due to its granular form from purchase. This made it difficult to compact into the crucible and gave to the need of a long degassing cycle which ultimately improved the deposition control.
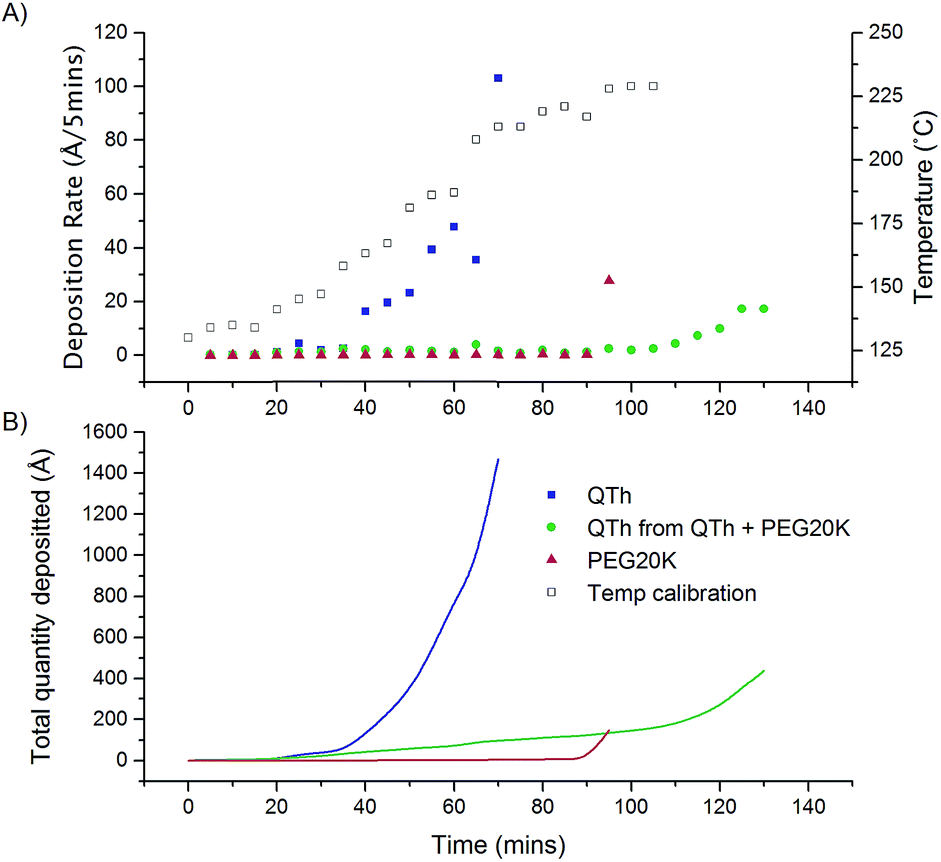 | ||
| Fig. 4 PVD rate studies as a function of time: (a) the first y axis shows the PVD deposition rate of QTh and QTh from PEG20K as well as a PEG baseline while the second y axis shows the temperature calibration curve, and (b) the cumulative deposition quantities on the QCM. | ||
The deposition rates as shown in Fig. 4A are a guideline, as it was found that the rates measured are a consequence of many factors including the rate at which depositions temperatures are approached and the length of the degassing phase. The time period of 15 minutes was selected as it allowed for the crucible to fully thermalize. When the rate was steady the deposition fluctuated between 0.1 and 0 Å s−1 however the total deposition rate levelled off to ∼0 Å s−1 over time within the 15 minute interval, before the next temperature increase. Each of the reported rate in Fig. 4A was an integration of the rate within that time interval. The stepwise increase in deposition temperature was performed until the melting temperature of the QTh (by which point explosive uncontrollable deposition of QTh would have occurred), however the PEG20K matrix prevents this from occurring. It should be noted that ∼400 Å were also deposited overnight during the degassing phase where the T was less than 130 °C from the blend and no such deposition occurred for the pure QTh (data not shown).
For the mixture of QTh + PEG20k higher rates were achieved when using intervals of less than 15 minutes; however a persistent rate of deposition for a single temperature was not achieved. In order to maintain appreciable deposition rates it was necessarily to continually increase the temperature. This is likely a consequence of the fact that deposition only occurs from the surface, and the process of deposition in the dispersion is a multistep process which involves dissolution of QTh into the PEG20K and then evaporation of dissolved QTh. This extraordinarily low rate of deposition allows for significantly higher control of the overall deposition quantity.
The use of the PEG blending technique commences deposition at well below the sublimation point for QTh and allows for significantly lower rates of deposition due to the fact that the vast majority of the QTh is held inside the PEG liquid, and sublimation can only occur from the PEG in the surface of that liquid. This allows for a new method of controlling the outcome of PVD processes. However this blending adds a different complexity to the process; PVD is usually performed by determining a constant deposition rate above the sublimation temperature, and then this is held until a desired quantity is reached. The blending method will not permit this as once deposition commences the rate is significantly lower, and the rate gradually reduces over time. Nevertheless this technique is employable and achieved extremely low deposition quantities.
This technique of adjusting the molecular packing is very simple and adaptable to other evaporants, can be used to adjust evaporation rates, and can provide significant benefits as it can overcome a range of existent processing issues. A key issue associated with using oxidants such as Fe(III)PTS is that the polymerisation temperature is restricted to temperatures at which the oxidant is a solid, as when it is a liquid, it will flow on the substrate, and will not form a uniform film. By removing the necessity to reach the melting temperature of the monomer, the processing temperature can be set at a value suited to the oxidant. However, care must be taken as some precursors, such as PEG may exhibit appreciable evaporation which can interfere with the polymerisation process. It can be recommended to use an IL as a component of the precursor solution, because even if the IL does evaporate, it is not interaction with the Fe(III) oxidant and can be washed away from the deposited film.
This technique can also be used to slow down evaporation rates of monomers as well, such that uncontrolled deposition does not occur when processing too far above the melting point; where the evaporation rate is very high. Situations like this can appear for monomers like pyrrole, where the melting point is at −23 °C and consequentially the deposition rate at room temperature can be uncontrollable.
For vacuum PVD process, using PEG20K blending is nontrivial and the technique can also allow for depositions of extremely low quantities.
Conclusions
A new versatile method for significantly broadening the operational range of physical vapour deposition and vapour phase polymerisation processes is outlined. By simply dispersing/dissolving an evaporant in an ionic liquid or high molecular weight polymer one can significantly increase the volatility due to the fact that the molecular ordering of the evaporant is, to an extent, broken. For VPP this process retains the purity associated with VPP processes for both the IL and high molecular weight polymer precursor. For PVD the deposition rate was significantly slowed while purity of the deposited medium was retained when using the high molecular weight evaporating medium.Notes and references
- T. Ishiguro, H. Kaneko, Y. Nogami, H. Ishimoto, H. Nishiyama, J. Tsukamoto, A. Takahashi, M. Yamaura, T. Hagiwara and K. Sato, Logarithmic Temperature Dependance of Resistivity in Heavily Doped Conducting Polymers at Low Temperature, Phys. Rev. Lett., 1992, 69, 660–663 CrossRef CAS PubMed.
- K. Lee, S. Cho, S. H. Park, A. J. Heeger, C.-W. Lee and S.-H. Lee, Metallic Transport in Polyaniline, Nature, 2006, 441, 65–68B CrossRef CAS PubMed.
- W. Jensen, D. Breiby and K. West, Base inhibited oxidative polymerisation of 3,4-ethylenedioxythiophene with iron(III)tosylate, Synth. Met., 2005, 152, 1–4 CrossRef.
- M. M. Szumilo, E. H. Gann, C. R. Mcneill, V. Lemaur, Y. Oliver, L. Thomsen, Y. Vaynzof, M. Sommer and H. Sirringhaus, Structure Influence on Charge Transport of Naphthalenediimide-Thiophene Copolymers, Chem. Mater., 2014, 26, 6796–6804 CrossRef CAS.
- W. E. Tenhaeff and K. K. Gleason, Initiated and Oxidative Chemical Vapor Deposition of Polymeric Thin Films: iCVD and oCVD, Adv. Funct. Mater., 2008, 18, 979–992 CrossRef CAS.
- J. S. Castrucci, J. D. Dang, B. A. Kamino, A. Campbell, D. Pitts, Z.-H. Lu and T. P. Bender, Considerations for the Physical Vapor Deposition of High Molar Mass Organic Compounds, Vacuum, 2014, 109, 26–33 CrossRef CAS.
- T. Engel and P. Reid, Physical Chemistry, Pearson, Boston, 3rd edn, 2013 Search PubMed.
- R. D. Rogers and K. R. Seddon, Ionic Liquids—Solvents of the Future?, Science, 2003, 302, 792–793 CrossRef PubMed.
- S. Lee and K. K. Gleason, Enhanced Optical Property with Tunable Band Gap of Cross-linked PEDOT Copolymers via Oxidative Chemical Vapor Deposition, Adv. Funct. Mater., 2015, 25, 85–93 CrossRef CAS.
- Ch. Kloc and R. Laudise, Vapor pressures of organic semiconductors: α-hexathiophene and α-quaterthiophene, J. Cryst. Growth, 1998, 193, 563–571 CrossRef CAS.
- B. Kolodziejczyk, D. Mayevsky and B. Winther-Jensen, Enhanced absorption spectra of conducting polymers co-polymerised from thiophene derivatives, RSC Adv., 2013, 3, 4568 RSC.
- K. Meerholz and J. Heinze, Electrochemical Solution and Solid-State Investigations on Conjugated Oligomers and Polymers of α-Thiophene and the p-Phenylene series, Electrochim. Acta, 1996, 41, 1839–1854 CrossRef CAS.
- Y. Furukawa, M. Akimoto and I. Harada, Vibration Key Bands and Electrical Conductivity of Polythiophene, Synth. Met., 1987, 18, 151–156 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Optical absorbance and XPS results of quaterthiophene. See DOI: 10.1039/c5ra16897j |
| This journal is © The Royal Society of Chemistry 2015 |