
Synthesis of new chiral lactam-fused pyridine derivatives†
Tetsuya Sengokua,
Yusuke Muratab,
Chihiro Suzukia,
Masaki Takahashia and
Hidemi Yoda*ab
aDepartment of Applied Chemistry, Faculty of Engineering, Shizuoka University, 3-5-1 Johoku, Naka-ku, Hamamatsu 432-8561, Japan. E-mail: tchyoda@ipc.shizuoka.ac.jp; Fax: +81 53 478 1150; Tel: +81 53 478 1150
bGraduate School of Science and Technology, Shizuoka University, 3-5-1 Johoku, Naka-ku, Hamamatsu 432-8561, Japan
First published on 25th August 2015
Abstract
New chiral C2-symmetric and unsymmetric lactam-fused pyridines have been synthesised in enantiomerically pure forms with satisfactory yields via an acid-promoted cyclisation of benzylidene-modified tetramic acids and various enamines.
Pyridine is a privileged structural motif in various medicinal and synthetic applications due to its broad utility as a bioactive agent,1 an effective ligand for organometallic catalysis2 and a functional unit for the fabrication of advanced organic materials.3 In this context, a considerable amount of research effort has been devoted to the development of synthetic chemistry of functionalised pyridines.4 Traditional approaches to the pyridine synthesis, as developed by Bohlmann–Rahtz,5 Hantzsch6 and Krönke,7 are based on cyclodehydration of carbonyl compounds with ammonia, which requires harsh conditions to offer poor functional group tolerance with respect to chemically labile substructures. Despite some efficient approaches developed during the past several years,8 success has generally been limited to just a few simple or achiral pyridine types, and therefore the challenge of accessing chiral complex structures still remain out of reach.
In the course of our synthetic studies concerning the use of amino acid-derived tetramic acids as chiral structural scaffolds,9 we have found that β-ketolactam moieties of these heterocyclic ring systems are capable of undergoing proline-catalysed tandem reactions of Knoevenagel condensation with aldehydes, followed by Michael addition, to generate a series of chiral diols 1 shown in Fig. 1.9d The intriguing structural architectures that would certainly find potential uses in preparing functional molecules have stimulated our synthetic interest to develop new types of pyridine ring systems featuring lactam-fused structures, so-called pyrrolo[3,4-b]pyridines, associated with C2-symmetric chiral motifs (2). An analogous class of molecular systems with the pyrrolo[3,4-b]pyridine nucleus, which have been shown to act at peripheral benzodiazepine receptors,10 were synthesised by Bhandari and his co-workers via lactam cyclisation of 2-aminomethyl-3-carboxypyridines with TFA. However, a challenging task suppressing epimerization11 at C3 stereocentre in the synthesis of chiral compounds still remained. Hence, it is a subject of great importance to develop an innovative methodology for their preparation, which will provide access to potential drug candidates. Here we report a new synthetic method for bislactam-fused pyridines possessing a C2-symmetric element in enantiomerically pure forms by using tetramic acid as a chiral source and its application to the synthesis of unsymmetric derivatives bearing a monolactam-fused pyridine core.
 | ||
| Fig. 1 Synthetic approaches toward chiral C2-symmetric lactam-fused pyridines 2. | ||
Our initial attempt to elaborate chiral diol 1 to the corresponding C2-symmetric lactam-fused pyridine 2 was to examine the cyclisation in the presence of an ammonium salt12 (Table S1, ESI†). Treatment of 1a (R1 = Bn, R2 = H) with an equimolar amount of ammonium acetate in AcOH at 100 °C led to an almost 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of pyridine derivatives, 2a and its epimer, in 62% yield, whereas no reaction occurred without heating (Table S1,† entries 1 and 2). On the other hand, using 1b (R1 = Bn, R2 = Et) in place of 1a resulted in decreased reaction efficiency, providing a diastereomeric mixture of 2b and epi-2b along with the corresponding dihydropyridine intermediate 3b (Table S1,† entry 3). The yield of 3b was dependent on the equivalents of ammonium acetate and the reaction time. In fact, the reaction of 1b with an excess amount of ammonium acetate (50 equiv.) completed within 1 h to afford 3b and its epimers (meso-isomers) in 62% combined yield with diastereomeric ratio of 35:65 (Table S1,† entry 4). The extensive screening of conditions revealed that the problematic epimerization occurred at high reaction temperatures (Table S1,† entries 5–7), despite the fact that the reaction at less than 60 °C gave no cyclised products. Attempts to avoid epimerization in the dihydropyridine formation with 1c also proved unsuccessful (Table S1,† entries 8 and 9). Thus, we turned our attention to find an alternative approach.
:1 mixture of pyridine derivatives, 2a and its epimer, in 62% yield, whereas no reaction occurred without heating (Table S1,† entries 1 and 2). On the other hand, using 1b (R1 = Bn, R2 = Et) in place of 1a resulted in decreased reaction efficiency, providing a diastereomeric mixture of 2b and epi-2b along with the corresponding dihydropyridine intermediate 3b (Table S1,† entry 3). The yield of 3b was dependent on the equivalents of ammonium acetate and the reaction time. In fact, the reaction of 1b with an excess amount of ammonium acetate (50 equiv.) completed within 1 h to afford 3b and its epimers (meso-isomers) in 62% combined yield with diastereomeric ratio of 35:65 (Table S1,† entry 4). The extensive screening of conditions revealed that the problematic epimerization occurred at high reaction temperatures (Table S1,† entries 5–7), despite the fact that the reaction at less than 60 °C gave no cyclised products. Attempts to avoid epimerization in the dihydropyridine formation with 1c also proved unsuccessful (Table S1,† entries 8 and 9). Thus, we turned our attention to find an alternative approach.
At this point, we assumed that the low reactivity of 1 toward dihydropyridine formation would be attributed to the inherent stability of the enol tautomer. Thus, we decided to investigate an intermolecular reaction of benzylidene tetramic acid 4 with tetramic acid-derived enamine 5 (Fig. 1). According to the reported procedure,13 a 4:1 mixture of geometric isomers of 4a was prepared by Knoevenagel condensation of a phenylalanine-derived tetramic acid with benzaldehyde in the presence of conc. HCl. Meanwhile, 5a was obtained in 54% yield by treatment with an excess amount of ammonium acetate at room temperature in MeOH.14 With these building blocks in hand, we attempted the reaction of 5a with 1.5 equivalents of 4a by stirring in MeOH, which led to complete consumption of 5a within 2 h. Subsequent addition of 1.5 equivalents of pyridinium p-toluenesulfonate (PPTS) followed by stirring at room temperature for 9 days gave 3c in low yield. This compound was aromatised upon treatment with MnO2 to afford 2c in 9% two-step yield from 5a (Table 1, entry 1). Although the product yield was quite low because of poor conversion to dihydropyridine intermediate, the enantiomeric purity of 2c proved to be higher than 99% ee. It should be noted that, even under reflux conditions in MeOH, 2c was given in optically pure form by this procedure (Table 1, entry 2). A further increase of the amount of PPTS to 3 equivalents could reduce the reaction time to 4 h, resulting in 45% yield of product, but failed to maintain the stereochemical integrity (93% ee) (Table 1, entry 3). With 0.5 equivalents of PPTS, the reaction also proceeded smoothly to provide 2c in 48% yield with >99% ee (Table 1, entry 4).
|
||||
|---|---|---|---|---|
| Entry | Additive [equiv.] | T | t [h] | 2c [% (% ee)] |
| a A solution of 4a and 5a in MeOH was stirred at room temperature for 2 h before the addition of PPTS.b Reactions were carried out in 1,2-dichloroethane.c Reaction was carried out in EtOH.d No reaction occurred. | ||||
| 1a | PPTS (1.5) | r.t. | 216 | 9 (>99) |
| 2a | PPTS (1.5) | Reflux | 6 | 41 (>99) |
| 3 | PPTS (3.0) | Reflux | 4 | 45 (93) |
| 4 | PPTS (0.5) | Reflux | 6 | 48 (>99) |
| 5 | Conc. HCl (0.5) | Reflux | 5 | 34 (98) |
| 6 | p-TsOH (0.5) | Reflux | 5 | 26 (>99) |
| 7 | AcOH (0.5) | Reflux | 6 | 44 (>99) |
| 8 | Malonic acid (0.5) | Reflux | 6 | 53 (>99) |
| 9 | Citric acid (0.5) | Reflux | 6 | 67 (>99) |
| 10b | ZnCl2 (0.5) | 70 °C | 6 | 0d (—) |
| 11b | BF3·OEt2 (0.5) | 70 °C | 6 | Trace (—) |
| 12c | Citric acid (0.5) | 70 °C | 6 | 58 (>99) |
Encouraged by the above results, we next screened additives to find the optimum conditions. The use of conc. HCl, p-TsOH15 or acetic acid was found ineffective under comparable conditions (Table 1, entries 5–7) and the reaction with Lewis acid, such as ZnCl2 and BF3·OEt2, failed to give the expected product (Table 1, entries 10 and 11). On the other hand, the reactions carried out with malonic acid and citric acid afforded the enantiopure product in higher yields of 53 and 67%, respectively (Table 1, entries 8 and 9). In continuing the efforts to improve the reaction efficiency, we used different solvents for comparison. The choice of EtOH resulted in decreased reaction efficiency and the reaction failed to reach full conversion within 6 h (Table 1, entry 12). In other polar solvents, such as t-BuOH, DMSO and H2O/EtOH (1/1), citric acid-mediated cyclisation proved ineffective. Consequently, we could identify the optimal conditions which involve the use of 0.5 equivalents of citric acid in refluxing MeOH.
Fig. S1a in the ESI† illustrates 1H NMR spectroscopic characteristics of 2c obtained with CDCl3 as a solvent. Pyridine 2c displays simple resonance patterns, indicating a genuine spectroscopic equivalence of the structural units. In addition, resonances assignable to the proton at the C3 position (Ha, δ 4.88 ppm) and one of benzyl protons (Hb, δ 3.75 ppm) were significantly shifted downfield relative to those of 4a (4.17 and 3.32 ppm, respectively). This behaviour indicated that these protons were located in the deshielding range of the newly formed pyridine ring. The symmetric nature of 2c was also shown in 13C NMR spectrum and resonances of all singlet carbons except for amide carbonyls could be assigned as aromatic carbons by DEPT experiment (Fig. S1b†). Furthermore, observed mass of 2c (446.1862, [M + H]+) was consistent with the molecular formula of C29H24N3O2 (calcd 446.1869). Thus, we unambiguously assigned the structure of 2c as depicted in Table 1.
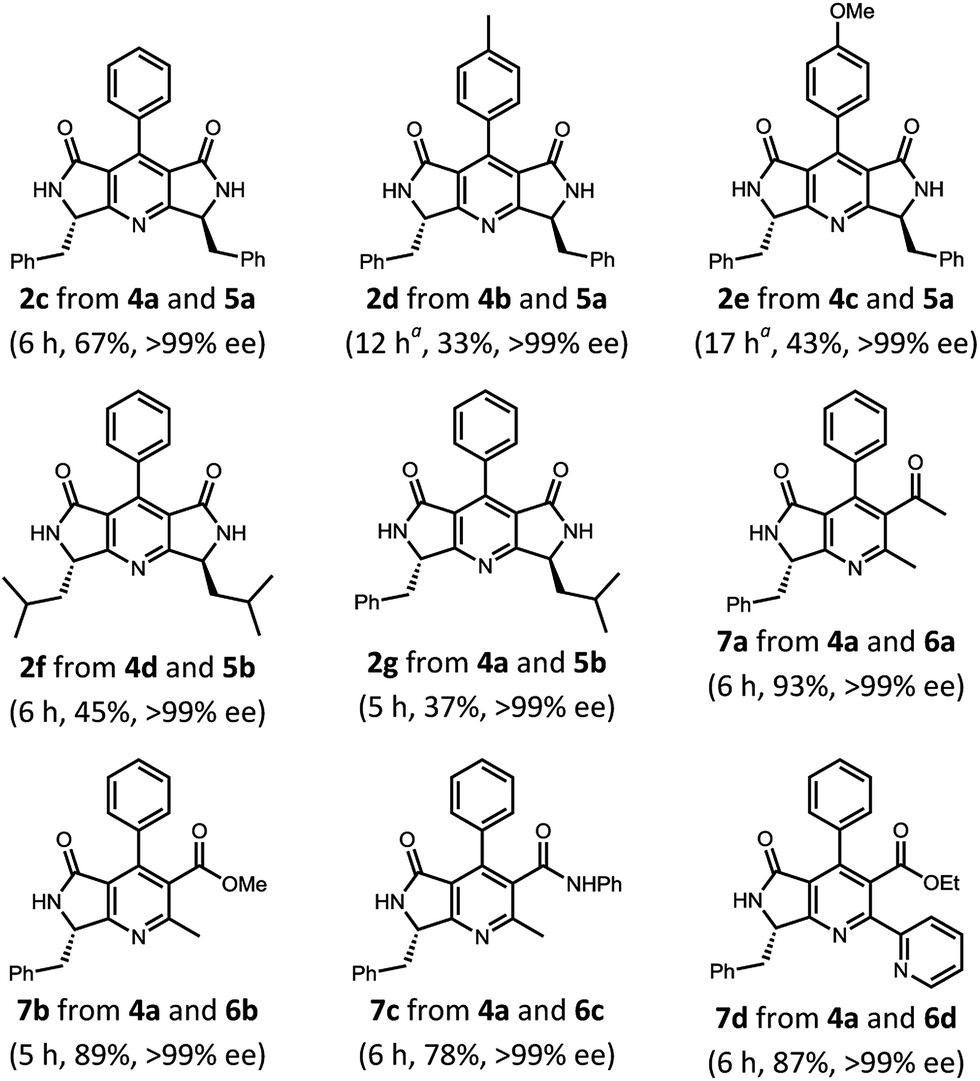
Table 2 shows the reactions of benzylidene tetramic acids 4 with various enamines (tetramic acid-derived enamines 5 and acyclic enamines 6). When p-tolyl derivative 4b was treated with 5a under the given conditions, the reaction proceeded sluggishly and gave 2d in 33% yield after 12 h in optically pure form.16 A similar reactivity was observed for substrate 4c, and 2e was obtained in 43% yield with >99% ee. In addition to the phenylalanine-derived pyrrolo[3,4-b]pyridines, leucine-derived pyridine 2f as well as phenylalanine–leucine mixed derivative 2g could be given in moderate yields (45 and 37%, respectively) also without loss of enantiopurity.17 The promising result of the productions of 2 channelled our interest to other substrate combinations in an attempt to prepare novel chiral unsymmetric lactam-fused pyridines. Along the line with the reactivity observed in the synthesis of 2, the reaction of 4a with 6a proceeded smoothly by refluxing for 6 h and gave the corresponding dihydropyridine as a diastereomeric mixture. These were subsequently oxidised to 7a in 93% yield without loss of stereochemical integrity. A favourable reactivity trend toward dihydropyridine formation was also observed for the cases of enamines 6b and c (prepared from methyl acetoacetate and acetoacetanilide), which led to give 7b and c in 89 and 78% yields, respectively, also in enantiomerically pure forms. Furthermore, the developed methodology was found to be applicable to more functionalised substrate 6d 18 bearing 2-pyridyl substituent, furnishing a single enantiomer of bipyridine derivative 7d in excellent yield.
|
|---|
| a A 1.5:1:0.5 mixture of 4, 5a and citric acid in MeOH was stirred at room temperature for 3 h before heating. |
 |

In conclusion, we have developed the new synthetic methodology for enantiopure C2-symmetric and unsymmetric lactam-fused pyridines. A key outcome of this work is citric acid-promoted cyclisation of benzylidene tetramic acids and various enamines. The new entry to these chiral pyridine derivatives will provide opportunities for future development of potential drug candidates.
Acknowledgements
We thank Dr Toshiyasu Inuzuka (Gifu University) for performing mass spectral analyses. We also thank Mr Yuki Nakamura and Mr Daichi Hayashi for partial support of this research. This work was supported financially in part by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan and the JGC-S Scholarship Foundation.Notes and references
- For recent examples, see: (a) M. Horiuch, C. Murakami, N. Fukamiya, D. Yu, T.-H. Chen, K. F. Bastow, D.-C. Zhang, Y. Takaishi, Y. Imakura and K.-H. Lee, J. Nat. Prod., 2006, 69, 1271 CrossRef CAS PubMed; (b) V. Onnis, T. T. Cocco, R. Fadda and C. Congiu, Bioorg. Med. Chem., 2009, 17, 6158 CrossRef CAS PubMed; (c) C. D. Duffy, P. Maderna, C. McCarthy, C. E. Loscher, C. Godson and P. J. Guiry, ChemMedChem, 2010, 5, 517 CrossRef CAS PubMed; (d) H. Motiwala, S. Kandre, V. Birar, K. S. Kadam, A. Rodge, R. D. Jadhav, M. Mahesh Kumar Reddy, M. K. Brahma, N. J. Deshmukh, A. Dixit, L. Doshi, A. Gupte, A. K. Gangopadhyay, R. A. Vishwakarma, S. Srinivasan, M. Sharma, K. V. S. Nemmani and R. Sharma, Bioorg. Med. Chem. Lett., 2011, 21, 5812 CrossRef CAS PubMed.
- (a) H. Nishiyama, H. Sakaguchi, T. Nakamura, M. Horihata, M. Kondo and K. Itoh, Organometallics, 1989, 8, 846 CrossRef CAS; (b) G. Desimoni, G. Faita and P. Quadrelli, Chem. Rev., 2003, 103, 3119 CrossRef CAS PubMed; (c) H.-L. Kwong, H.-L. Yeung, C.-T. Yeung, W.-S. Lee, C.-S. Lee and W.-L. Wong, Coord. Chem. Rev., 2007, 251, 2188 CrossRef CAS PubMed.
- (a) V. C. Gibson, C. Redshaw and G. A. Solan, Chem. Rev., 2007, 107, 1745 CrossRef CAS; (b) S.-J. Su, T. Chiba, T. Takeda and J. Kido, Adv. Mater., 2008, 20, 2125 CrossRef CAS PubMed.
- (a) G. D. Henry, Tetrahedron, 2004, 60, 6043 CrossRef CAS PubMed; (b) E. F. V. Scriven, in Pyridines: from lab to production, Academic Press, Oxford, UK, 1st edn, 2013 Search PubMed.
- F. Bohlmann and D. Rahtz, Chem. Ber., 1957, 90, 2265 CrossRef CAS PubMed.
- A. Hantzsch, Liebigs Ann. Chem., 1882, 215, 1 CrossRef PubMed.
- (a) W. Zecher and F. Kröhnke, Chem. Ber., 1961, 94, 690 CrossRef CAS PubMed; (b) F. W. Zecher and F. Kröhnke, Chem. Ber., 1961, 94, 698 CrossRef PubMed.
- For selected examples, see: (a) Y. S. Chun, J. H. Lee, J. H. Kim, Y. O. Ko and S. Lee, Org. Lett., 2011, 13, 6390 CrossRef CAS PubMed; (b) S. Yamamoto, K. Okamoto, M. Murakoso, Y. Kuninobu and K. Takai, Org. Lett., 2012, 14, 3182 CrossRef CAS PubMed; (c) L. Zheng, J. Ju, Y. Bin and R. Hua, J. Org. Chem., 2012, 77, 5794 CrossRef CAS PubMed; (d) C. Allais, F. Liéby-Muller, J. Rodriguez and T. Constantieux, Eur. J. Org. Chem., 2013, 2013, 4131 CrossRef CAS PubMed; (e) X. He, Y. Shang, Z. Yu, M. Fang, Y. Zhou, G. Han and F. Wu, J. Org. Chem., 2014, 79, 8882 CrossRef CAS PubMed; (f) L. Luo, L. Meng, Y. Peng, Y. Xing, Q. Sun, Z. Ge and R. Li, Eur. J. Org. Chem., 2015, 2015, 631 CrossRef CAS PubMed.
- (a) T. Sengoku, J. Wierzejska, M. Takahashi and H. Yoda, Synlett, 2010, 2944 CAS; (b) T. Sengoku, Y. Nagae, Y. Ujihara, M. Takahashi and H. Yoda, J. Org. Chem., 2012, 77, 4391 CrossRef CAS PubMed; (c) Y. Ujihara, K. Nakayama, T. Sengoku, M. Takahashi and H. Yoda, Org. Lett., 2012, 14, 5142 CrossRef CAS PubMed; (d) T. Sengoku, K. Suzuki, K. Nakayama, F. Yagishita, M. Sakamoto, M. Takahashi and H. Yoda, RSC Adv., 2014, 4, 30775 RSC.
- M. Anzini, A. Cappelli, S. Vomero, G. Giorgi, T. Langer, G. Bruni, M. R. Romeo and A. S. Basile, J. Med. Chem., 1996, 39, 4275 CrossRef CAS PubMed.
- A. Bhandari, B. Li and M. A. Gallop, Synthesis, 1999, 1951 CrossRef CAS.
- M. Augustin and P. Jeschke, J. Prakt. Chem., 1987, 329, 985 CrossRef CAS PubMed.
- K. Matsuo and K. Tanaka, Yakugaku Zasshi, 1984, 104, 1004 CAS.
- N. R. Gade, V. Devendram, M. Pal and J. Iqbal, Chem. Commun., 2013, 49, 7926 RSC.
- (a) K. Matsuo, M. Adachi and T. Takagi, Chem. Express, 1992, 7, 465 CAS; (b) K. Matsuo, M. Adachi, T. Takagi, S. Ueno, T. Arase and F. Kuroi, Heterocycles, 2005, 65, 377 CrossRef CAS.
- When the reaction of 4b with 5a was carried out for 17 h, 93% enantiomeric excess of 2d was observed.
- These products were revealed to be thermodynamically susceptible, partially epimerising at room temperature. For example, the enantiomeric excess of 2f stored for 10 days at room temperature was only 60%.
- J. H. Lee, B. S. Choi, J. H. Chang, H. B. Lee, J.-Y. Yoon, J. Lee and H. Shin, J. Org. Chem., 2007, 72, 10261 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterisation data, 1H and 13C NMR spectra for the compounds 2, 4, 5 and 7. See DOI: 10.1039/c5ra16896a |
| This journal is © The Royal Society of Chemistry 2015 |