
Ni(II) complexes bearing pyrrolide-imine ligands with pendant N-, O- and S-donor groups: synthesis, structural characterization and use in ethylene oligomerization†
A. C.
Pinheiro
a,
A. H.
Virgili
a,
T.
Roisnel
b,
E.
Kirillov
c,
J.-F.
Carpentier
*c and
O. L.
Casagrande
Jr.
*a
aLaboratory of Molecular Catalysis, Instituto de Química, Universidade Federal do Rio Grande do Sul, Avenida Bento Gonçalves, 9500, RS 90501-970, Brazil. E-mail: osvaldo.casagrande@ufrgs.br
bInstitut des Sciences Chimiques de Rennes, Centre de diffraction X, UMR 6226 CNRS-Université de Rennes 1, F-35042 Rennes Cedex, France
cInstitut des Sciences Chimiques de Rennes, Organometallics: Materials and Catalysis Dept., UMR 6226 CNRS-Université de Rennes 1, F-35042 Rennes Cedex, France. E-mail: jean-francois.carpentier@univ-rennes1.fr
First published on 15th October 2015
Abstract
A series of new Ni(II) complexes of general formula {L}NiCl [Ni1, L = 2-(C4H3N-2′-CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N)C2H4NHPh; Ni2, L = 5-tert-butyl-2-(C4H2N-2′-CHN)C2H4NHPh; Ni3, L = 2-(C4H3N-2′-CHN)C2H4OPh; Ni4, L = 2-(C4H3N-2′-CHN)C6H4-2′-OPh; Ni5, L = 2-(C4H3N-2′-CHN)C6H4-2′-SPh; Ni6, L = 2-(C4H3N-2′-CHN)CH2C6H4-2′-OMe] were prepared and fully characterized. All nickel precatalysts, activated with methylaluminoxane (MAO), exhibited moderate to good activities for ethylene oligomerization [TOF = 6.1–71.3 × 103 mol (C2H4) (mol (Ni)−1 h−1)] and producing high selectivities for 1-butene (68.3–94.0 wt%). The catalytic performance was substantially affected by the ligand environment regarding the pendant oxygen- and sulfur-donor groups, and the substituents on the pyrrolide group. Activation of nickel precatalyst Ni3 with ethylaluminum sesquichloride (Et3Al2Cl3, EASC) instead of MAO produced a significantly more productive catalyst system than Ni2/MAO (TOF = 153
N)C2H4NHPh; Ni2, L = 5-tert-butyl-2-(C4H2N-2′-CHN)C2H4NHPh; Ni3, L = 2-(C4H3N-2′-CHN)C2H4OPh; Ni4, L = 2-(C4H3N-2′-CHN)C6H4-2′-OPh; Ni5, L = 2-(C4H3N-2′-CHN)C6H4-2′-SPh; Ni6, L = 2-(C4H3N-2′-CHN)CH2C6H4-2′-OMe] were prepared and fully characterized. All nickel precatalysts, activated with methylaluminoxane (MAO), exhibited moderate to good activities for ethylene oligomerization [TOF = 6.1–71.3 × 103 mol (C2H4) (mol (Ni)−1 h−1)] and producing high selectivities for 1-butene (68.3–94.0 wt%). The catalytic performance was substantially affected by the ligand environment regarding the pendant oxygen- and sulfur-donor groups, and the substituents on the pyrrolide group. Activation of nickel precatalyst Ni3 with ethylaluminum sesquichloride (Et3Al2Cl3, EASC) instead of MAO produced a significantly more productive catalyst system than Ni2/MAO (TOF = 153![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 vs. 43500 mol (C2H4) (mol (Ni)−1 h−1)); however, the 1-butene selectivity was drastically reduced, attaining only 53 wt% with a concomitant production of larger amounts of internal butenes (38 wt%). Under optimized conditions ([Ni] = 10 μmol, 30 °C, oligomerization time = 20 min, 20 bar ethylene, [Al]/[Ni] = 250), precatalyst Ni3 led to a TOF = 55900 mol (C2H4) (mol (Ni)−1 h−1) and 82.8 wt% selectivity for 1-butene.
700 vs. 43500 mol (C2H4) (mol (Ni)−1 h−1)); however, the 1-butene selectivity was drastically reduced, attaining only 53 wt% with a concomitant production of larger amounts of internal butenes (38 wt%). Under optimized conditions ([Ni] = 10 μmol, 30 °C, oligomerization time = 20 min, 20 bar ethylene, [Al]/[Ni] = 250), precatalyst Ni3 led to a TOF = 55900 mol (C2H4) (mol (Ni)−1 h−1) and 82.8 wt% selectivity for 1-butene.
Introduction
The oligomerization of ethylene is one of the most important industrial processes to obtain linear α-olefins (LAOs).1 LAOs are extensively used for preparing detergents, lubricants, plasticizers, and oil field chemicals or used as co-monomers, etc.2 Among the different classes of catalysts used for the production of α-olefins, nickel complexes containing P,P-,3 P,N-,4 P,O-,5 N,N-,6 or N,O-7 bidentate chelating ligands are the most frequently studied. More recently, nickel complexes bearing tridentate ligands have attracted much interest owing to their good to excellent performance towards the production of α-olefins.8 In this context, many different classes of tridentate ligands bearing a variety of E-donor groups (E = N, O, P, and S) have been explored and used to support nickel complexes (Chart 1). In particular, Braunstein et al. reported the use of a tridentate N,P,N-type ligand in the synthesis of the pentacoordinated mononuclear nickel complex [NiCl2(NOPONMe2–N,P,N)] (NOPONMe2 = bis(4,4-dimethyl-2-oxazolyldimethylmethoxy)phenylphosphine), which is an effective precatalyst leading to selectivities in C4 olefins higher than 90% when activated with ethylaluminum dichloride (EtAlCl2).9 Sun et al. described the synthesis of nickel complexes bearing 2-(benzimidazol-2-yl)-1,10-phenanthrolines; upon activation with diethylaluminum chloride (Et2AlCl), high catalytic activity of up to 1.27 × 107 g mol (Ni)−1 h−1 and high selectivity for 1-butene (90.5 wt%) could be achieved.10 More recently, Olivier-Bourbigou et al. disclosed a new class of nickel complexes based on imino-imidazole ligands bearing a pendant donor group that are able to oligomerize ethylene in presence of EtAlCl2 or methylaluminoxane (MAO), producing mostly dimers and trimers.11 | ||
| Chart 1 Examples of nickel complexes based on tridentate ligands applied in ethylene oligomerization. | ||
In recent years our groups have been interested in exploring the potential applications of tridentate ligands in the oligomerization catalysis field.12 Thus, we have previously reported on the use of nickel complexes based on tridentate nitrogen-, oxygen- or sulfur-bridged bis(pyrazolyl) ligands as highly selective and productive precatalysts for ethylene dimerization upon activation with MAO.12a Replacement of a pyrazole group by a pendant ether or thioether group on one side of the ether-bis(pyrazolyl) ligand led to more active systems.12f
As a continuation of the work outlined above, we report here the synthesis and structural characterization of a new family of Ni(II) complexes bearing pyrrole-imine ligands with pendant O- and S-donor groups and their catalytic behavior for ethylene oligomerization upon activation with MAO and ethylaluminum sesquichloride (Et3Al2Cl3, EASC). We discuss the performance of these catalysts, evaluating the role of the ligand, and the experimental parameters on the activity and selectivity towards the production of 1-butene.
Results and discussion
Synthesis and characterization of Ni(II) complexes bearing pyrrolide-imine ligands with pendant N-, O- and S-donor groups
The pyrrole-imino pro-ligands with pendant N-, O- and S-donor groups were readily synthesized by Schiff base condensations between the corresponding primary amines and pyrrole-2-carboxaldehyde in refluxing methanol. The identity of this class of ligands was established by IR and NMR spectroscopy, elemental analysis, and by an X-ray diffraction study for ligands L3, L4 and L5 (see ESI†). Treatment of L1–L6 with 1.0 equiv. of NaH and then NiCl2(dme) yielded the corresponding nickel complexes {L}NiCl (Ni1–Ni6), which were isolated as brown solids in moderate to good yields (63–90 wt%) (Scheme 1). These nickel complexes show moderate solubility in acetonitrile or THF at room temperature. The 1H NMR spectra of these complexes featured very broad resonances and proved uninformative. As complexes of Ni(II) with square-planar geometry are expected to be diamagnetic, this observation implies that these complexes are distorted from the ideal geometry in solution. Similar results have been found for salicylaldiminato nickel(II) halide complexes.13 The identity of Ni1–Ni6 was established on the basis of elemental analysis and single-crystal X-ray diffraction studies for Ni1. | ||
| Scheme 1 | ||
Single crystals of Ni1 suitable for X-ray diffraction were grown from a concentrated acetonitrile/ether solution (80:20) at room temperature. In the solid state, this complex is monomeric with κ3 coordination of the monoanionic pyrrolide-imine–amine ligand onto the nickel center. As shown in Fig. 1, Ni1 adopts a distorted square planar geometry around nickel(II). The N(1), N(7), N(10), and Cl(1) atoms are nearly coplanar, with Cl and N(7) occupying the trans positions (Cl(1)–Ni(1)–N(7) = 179.04(5)°, N(1)–Ni(1)–N(10): 168.77(6)°). The Ni(1)–N(1) and Ni(1)–N(7) bond distances [1.8715(4) Å, 1.8585(14) Å, respectively] are close to the values previously reported for Ni(II) complexes having pyrrole-imine ligands.14
 | ||
| Fig. 1 ORTEP drawing of Ni1. Ellipsoids are drawn at the 50% probability level. Selected bond distances (Å) and angles (deg): Ni(1)–Cl(1) = 2.1774(4), Ni(1)–N(1) = 1.8715(14), Ni(1)–N(7) = 1.8585(14), Ni(1)–N(10) = 1.9335(14). N(7)–Ni(1)–Cl(1) = 179.04(5). N(1)–Ni(1)–N(10) = 168.77(6), N(7)–Ni(1)–N(1) = 83.74(6), N(7)–Ni(1)–N(10) = 85.37(6), Cl(1)–Ni(1)–N(10) = 94.04(4), N(1)–Ni(1)–Cl(1) = 96.81(4). | ||
Ethylene oligomerization studies
The performance of nickel precatalysts Ni1–Ni6 were explored in ethylene oligomerization using as cocatalyst methylaluminoxane (MAO) containing 20 wt% AlMe3, at 30 °C and 20 bar constant ethylene pressure. Table 1 summarizes the results of reactions carried out using 10 μmol of precatalyst in 40 mL of toluene. All experiments were at least duplicated, yielding reproducible results within ±10%. When activated with MAO, all the nickel complexes investigated were found to generate active systems for the production of short-chain olefins in the C4–C6 range with turnover frequencies (TOFs) varying from 6100 to 71300 mol (C2H4) mol (Ni)−1 h−1. As shown in Table 1 (entries 1–6), the ligand environment regarding the pendant oxygen- and sulfur-donor groups, and the substituents on the pyrrolide group influenced the catalytic performance of the nickel precatalysts on ethylene oligomerization, as can be better visualized in Fig. 2. It is should be pointed out that, taking into account the lower donor ability of the ether- and thio- groups as compared to the amine unit, we cannot rule out that L3–L6 could act as bidentate and not tridentate ligands. In the absence of structural data for Ni3–Ni6, we can only speculate at this stage that the coordination of these pendant oxygen- and sulfur-donor groups onto the nickel center could stabilize the active catalytic species.
| Entry | Cat | [Al]/[Ni] | Olig. (g) | TOFb (×103) | Selectivity (wt%) | |
|---|---|---|---|---|---|---|
| C4 (α-C4) | C6 (α-C6) | |||||
| a Reaction conditions: toluene = 40 mL, [Ni] = 10 μmol, oligomerization time = 20 min, P(ethylene) = 20 bar (kept constant), Tpol = 30 °C. The results shown are representative of at least duplicated experiments. b Mol of ethylene converted (mol of Ni)−1 h−1 as determined by quantitative GLC. c T = 50 °C. d Oligomerization time = 5 min. e Oligomerization time = 40 min. f EASC as cocatalyst. | ||||||
| 1 | Ni1 | 250 | 1.10 | 11.6 | 97.2 (92) | 2.8 (42) |
| 2 | Ni2 | 250 | 2.90 | 29.1 | 97.1 (83) | 2.9 (43) |
| 3 | Ni3 | 250 | 4.00 | 43.5 | 97.4 (85) | 2.6 (30) |
| 4 | Ni4 | 250 | 1.40 | 14.7 | 98.9 (84) | 1.1 (30) |
| 5 | Ni5 | 250 | 7.10 | 71.3 | 94.8 (72) | 5.2 (22) |
| 6 | Ni6 | 250 | 0.60 | 6.10 | 100 (94) | 0.0 |
| 7 | Ni3 | 500 | 1.00 | 11.0 | 97.7 (90) | 2.3 (52) |
| 8 | Ni3 | 100 | 2.50 | 26.6 | 98.2 (90) | 1.8 (44) |
| 9 | Ni3 | 50 | 0.50 | 2.10 | 97.7 (91) | 2.3 (71) |
| 10c | Ni3 | 250 | 2.50 | 27.3 | 97.4 (85) | 2.6 (33) |
| 11d | Ni3 | 250 | 1.40 | 55.9 | 96.4 (86) | 3.6 (49) |
| 12e | Ni3 | 250 | 3.50 | 18.9 | 94.5 (79) | 5.5 (29) |
| 13f | Ni3 | 50 | 15.3 | 153.7 | 91.5 (58) | 8.5 (17) |
 | ||
| Fig. 2 Influence of the nature of nickel precatalyst on TOF and selectivity for 1-butene (T = 30 °C, 20 bar, time = 20 min, [Al]/[Ni] = 250). | ||
Precatalyst Ni1, when activated with 250 equiv. of MAO, was found to give moderate activity [TOF = 11600 mol (C2H4) mol (Ni)−1 h−1] along with good selectivity towards 1-butene (89 wt%). Increasing the steric hindrance on the pyrrolide unit, as in precatalyst Ni2, caused an increase in activity by a factor of 2.50 suggesting that the tert-butyl substituent acts as a protecting group of the active species, thereby increasing the catalytic productivity.6b,15 Replacing the amine with an ether group as in precatalyst Ni3 led to a much higher activity [TOF = 43500 mol (C2H4) mol (Ni)−1 h−1] as compared to the counterpart Ni1 [TOF = 11600 mol (C2H4) mol (Ni)−1 h−1]; however, a slight decreasing in 1-butene selectivity was found using Ni3 [82.8 wt% for Ni3vs. 89.4 wt% for Ni1].
The productivity for ethylene oligomerization is also substantially affected by the ligand framework. Hence, precatalyst Ni3 that contains an ethylene bridge unit is ca. 3.0 times more active than Ni4 that contains a more rigid phenyl moiety (compare entries 3 and 4). This result possibly arises from the presence of a electron-withdrawing group (phenyl unit) that decreases the Lewis base of the ether unit (OPh), and thus destabilizing the Ni(II) active species. On the other hand, the selectivity for butenes and especially 1-butene were similar (ca. 83.0 wt%), indicating that this variation in the ligand structure had no influence on the product distribution.
Replacement of the ether by a thioether donor group (Ni5) led to a highly active oligomerization system (TOF = 71300 mol (C2H4) mol (Ni)−1 h−1) associated to a good selectivity for 1-butene production (68.3 wt%). Under identical oligomerization conditions (10 μmol [Ni], 30 °C, 20 bar of ethylene, [Al]/[Ni] = 250, time = 20 min) Ni5 is much more active than similar nickel complexes such as NiCl2{bis[2-(3,5-dimethylpyrazolyl)ethyl)]ether} [TOF = 7100 mol (C2H4) mol (Ni)−1 h−1]12a and NiCl2{1-(2-(2-phenoxyethoxy)ethyl)-3,5-dimethyl-1H-pyrazole} [TOF = 35800 mol (C2H4) mol (Ni)−1 h−1].12f As suggested above we assume that this higher productivity may arise from the softer and greater donor ability of S (as compared to O-donor) leading to substantially improved catalyst lifetimes. Similar results have been found for the class of nickel precatalysts NiCl2(NZN) based on nitrogen-, oxygen-, or sulfur bridged ligands, whereas the replacement of O-bridged donor atom by S- promotes a much higher activity for NiCl2{bis[2-(3,5-dimethylpyrazolyl)ethyl)]sulfide} [TOF = 57200 mol (C2H4) mol (Ni)−1 h−1] as compared to NiCl2{bis[2-(3,5-dimethylpyrazolyl)ethyl)]ether} [TOF = 7100 mol (C2H4) mol (Ni)−1 h−1].12a
When the length of the spacer between the central imine and the ether donor group was increased by one carbon (compare complexes Ni6 with Ni3 and Ni4), the productivity decreased substantially [TOF = 6100 mol (C2H4) mol (Ni)−1 h−1]. In this case, we surmise a more difficult coordination of the OMe group onto the metal center to deliver a stable 6-membered-ring nickel complex.
The preliminary study was extended to investigate the effect of temperature, [Al]/[Ni] molar ratio, cocatalyst type, and oligomerization time on the catalytic performance of the more promising system, i.e.Ni3/MAO (Table 1, entries 7–13). Elevating the temperature from 30 °C to 50 °C led to a reduction in productivity [TOF = 27300 mol (C2H4) mol (Ni)−1 h−1], suggesting that a partial decomposition of the active catalytic species took place. However, it should be pointed out that even at higher temperature (50 °C), the Ni3/MAO still operates with high activity and good selectivity for 1-C4 production (82.8 wt%) when compared to similar nickel precatalysts bearing tridentate ligands.8
The catalyst selectivities and activities were monitored as a function of time in the ethylene oligomerization reaction promoted by the Ni3/MAO catalytic system (Fig. 3). The oligomerization activity progressively decreased when the reaction time was prolonged from 5 to 20 min and finally to 40 min; this indicates that the catalyst lifetime is relatively short. Furthermore, it is important to note that Ni3 undergoes almost complete deactivation after 20 min. On the other hand, the reaction time causes a minimum impact on the selectivity for 1-butene, varying from 82.9 wt% (5 min) to 74.6 wt% (40 min), indicating that a single species is operative in this system. These results also indicate that isomerization of butenes is a minor pathway in these reactions and that the observed selectivities directly reflect the dimerization abilities of the catalyst.
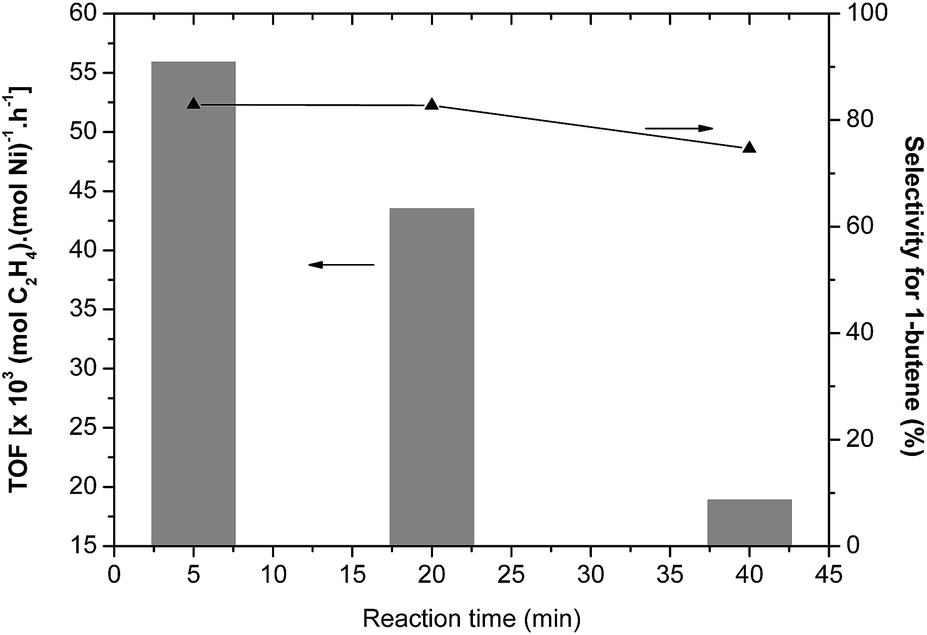 | ||
| Fig. 3 Monitoring of selectivities and activities as a function of time in the ethylene oligomerization reaction promoted by the Ni3/MAO system (T = 30 °C, 20 bar of ethylene, [MAO]/[Ni] = 250). | ||
The influence of the MAO loading on the catalyst behavior was also studied. When activated with 50 equiv. of MAO, precatalyst Ni3 gave a lower activity [TOF = 2100 mol (C2H4) mol (Ni)−1 h−1, entry 9], which was increased upon using 100 equiv. [TOF = 26600 mol (C2H4) mol (Ni)−1 h−1, entry 8] and even further with 250 equiv. [TOF = 43500 mol (C2H4) mol (Ni)−1 h−1, entry 3]. On the other hand, a greater loading of MAO (500 equiv.) led to lower activity (entry 7). In all cases, the use of different MAO loading ([Al]/[Ni] = 50–500) had little impact on the selectivity for α-C4 that remained in the range of 82.8–88.4 wt%.
Activation of the nickel precatalyst Ni3 with 50 equiv. of ethylaluminum sesquichloride (Et3Al2Cl3, EASC) instead of MAO produced a significantly better catalyst system with TOF = 153700 mol (C2H4) mol (Ni)−1 h−1. This high activity eventually generated some exothermicity, so that the reaction with Ni3/EASC performed at an initial temperature of 30 °C (entry 14) rapidly rose to 40–45 °C. Although the catalyst activity was improved by approximately 75-fold (compare entry 9 vs. 13), the 1-butene selectivity was drastically reduced, dropping down to 58 wt% with a concomitant production of larger amounts of internal butenes (33.5 wt%) and hexenes (8.5 wt%). The possibility of using small amounts of cocatalyst is obviously the most interesting feature of this catalyst system.
Conclusions
A new set of tetracoordinate nickel complexes bearing pyrrolide-imine ligands with pendant N-, O- and S-donor groups were synthesized and structurally characterized. Upon activation with MAO or EASC co-catalysts, these Ni(II) complexes showed moderate to high catalytic activity for ethylene oligomerization with good selectivity towards 1-butene production. Variations of the ligand structure demonstrated that a dramatic change in catalytic behavior can be obtained upon a subtle modification in the ligand skeleton. For instance, with nickel complex chelated by L2, the increase in activity suggested the beneficial role of the tert-butyl group as sterically protecting group of the active species. On the other hand, an expansion of the imino-ether arm by introduction of one additional CH2 unit in the spacer, generating a likely more flexible ligand L6, decreased substantially the activity of Ni6; this can be tentatively associated to the formation of a less stable 6-membered-ring nickel complex. Within the prepared series, the most active nickel catalyst (Ni5) is the one with a more rigid ligand bearing a soft donor-group (SPh). However, variation of the ligand structure does not play a significant role in the selectivity for butenes and especially 1-butene, attaining 72.0–94.0 wt% of the total amount of olefins formed in the oligomerization reactions. The use of EASC instead of MAO afforded a highly active species; however the 1-butene selectivity was drastically reduced.Experimental
General procedures
All manipulations involving air- and/or water-sensitive compounds were carried out in an MBraun glovebox or under dry argon using standard Schlenk techniques. Solvents were dried from the appropriate drying agents under argon before use. The ligands 2-(C4H4N-2′-CHN)C2H4(NH)Ph (L1), 5-tert-butyl-2-(C4H3N-2′-CHN)C2H4(NH)Ph (L2), and 2-(C4H4N-2′-CHN)C2H4OPh (L3) were prepared by literature procedures.16 5-tert-Butyl-1H-pyrrole-2-carboxyaldehyde was prepared using the reported procedure.17 NiCl2(dme), pyrrole-2-carboxyaldehyde, 2-(phenylthio)aniline, 2-phenoxyaniline, and 2-methoxybenzylamine were purchased from Sigma-Aldrich and used as received. Ethylene (White Martins Co.) and argon were deoxygenated and dried through BTS columns (BASF) and activated molecular sieves prior to use. MAO (Witco, 5.21 wt% Al solution in toluene) was used as received. EASC (Akzo Nobel) was used with the previous dilution (2.1 wt% Al solution in toluene). Infrared spectra were performed on neat products using a FT-IR Bruker Alpha Spectrometer operating in the ATR mode. 1H and 13C{1H} NMR spectra were recorded at 25 °C on a Varian Inova 300 spectrometer operating at 300 MHz. Chemical shifts are reported in ppm vs. SiMe4 and were determined by reference to the residual solvent peaks. Elemental analyses were performed by the Analytical Central Service of the Institute of Chemistry-USP (Brazil) and are the average of two independent determinations. Quantitative gas chromatographic analysis of ethylene oligomerization products was performed on a Agilent 7890A instrument equipped with a Petrocol HD capillary column (methyl silicone, 100 m length, 0.25 mm i.d. and film thickness of 0.5 μm) (36 °C for 15 min, then heating at 5 °C min−1 until 250 °C); cyclohexane was used as internal standard.
Synthesis of the pyrrole-imine ligands with O- and S-donor groups
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) N)Ph-2-OPh (L4).
To a stirred solution containing pyrrole-2-carboxyaldehyde (0.250 g, 2.63 mmol) in ethanol (75 mL), 2-phenoxyaniline (0.560 g, 2.63 mmol) was added. The reaction mixture was stirred for 72 h at 65 °C. The solvent was evaporated to give a pale brown solid that was recrystallized from ethanol/ether to give L4 as an off-white solid (0.413 g, 87%). Mp: 110.4 °C. 1H NMR (300 MHz, CDCl3, 25 °C): δ 6.21 (dd, 2J = 2.6 and 3.6 Hz, 1H, H-pyrrole), 6.60 (dd, 2J = 1.3 and 3.6 Hz, 1H, H-py), 6.79 (d, 2J = 1.3 Hz, 1H, H-pyrrole), 6.90–7.27 (m, 9H, Ar–H), 8.23 (s, 1H, CHN), 9.69 (br s, 1H, NH-pyrrole). 13C NMR (125 MHz, CDCl3, 25 °C): δ 110.38 (CH, 4-pyrrole), 116.68 (CH, 3-pyrrole), 117.89 (2CH, Ar–C), 120.98 (CH, 5-pyrrole), 121.20 (CH, Ar–C), 122.56 (CH, Ar–C), 123.31 (CH, Ar–C), 124.82 (CH, Ar–C), 126.25 (CH, Ar–C), 129.61 (2CH, Ar–C), 130.98 (C, 2-pyrrole), 144.04 (C, Ar–C), 149.21 (C, Ar–C), 150.88 (CH, NC–H), 158.27 (C, Ar–C). IR (KBr, cm−1) ν: 3238 (s), 3095 (w), 3063 (w), 2979 (w), 2896 (w), 1687 (m), 1631 (s), 1594 (m), 1488 (s), 1455 (m), 1418 (m), 1339 (w), 1319 (w), 1267 (w), 1237 (s), 1205 (m), 1184 (w), 1159 (s), 1134 (m), 1090 (m), 1069 (w), 969 (w), 940 (w), 878 (m), 845 (m), 795 (m), 751 (s), 693 (m), 603 (m), 490 (w), 460 (w). Anal. calcd for C17H14N2O: C, 77.84; H, 5.38; N, 10.68. Found: C, 77.26; H, 5.09; N, 10.69.
N)CH2Ph-2-SPh (L5).
This product was prepared as described above for L4, starting from pyrrole-2-carboxyaldehyde (0.250 g, 2.63 mmol) and 2-(phenylthio)aniline (0.529 g, 2.63 mmol). L5 was recovered as an off-white solid (0.219 g, 30%). Mp: 102.5 °C. 1H NMR (300 MHz, CDCl3, 25 °C): δ 6.30 (dd, 2J = 2.7 and 3.5 Hz, 1H, H-pyrrole), 6.67 (dd, 2J = 1.1 and 3.6 Hz, 1H, H-pyrrole), 6.96–7.07 (m, 4H, H–Ar + H-pyrrole), 7.16–7.22 (m, 1H, H–Ar), 7.30–7.37 (m, 3H, H–Ar), 7.44–7.47 (m, 2H, H–Ar), 8.16 (s, 1H, CHN), 9.55 (br s, 1H, NH-pyrrole). 13C NMR (125 MHz, CDCl3, 25 °C): δ 110.55 (CH, 4-pyrrole), 116.70 (CH, 3-pyrrole), 118.42 (CH, 5-pyrrole), 123.38 (CH, Ar–C), 125.88 (CH, Ar–C), 127.14 (CH, Ar–C), 127.84 (CH, Ar–C), 128.93 (2CH, Ar–C), 129.37 (2CH, Ar–C), 130.93 (CH, Ar–C), 132.46 (C, 2-pyrrole), 133.46 (C, Ar–C), 134.19 (C, Ar–C), 149.77 (CH, NC–H), 149.87 (C, Ar–C). IR (KBr, cm−1) ν: 3215 (m), 3165 (w), 3053 (w), 2975 (w), 2906 (w), 2864 (w), 1615 (s), 1562 (s), 1551 (m), 1463 (m), 1438 (m), 1405 (s), 1339 (m), 1315 (m), 1302 (w), 1262 (m), 1242 (w), 1198 (s), 1131 (s), 1088 (s), 1058 (m), 1032 (s), 962 (w), 935 (w), 912 (w), 879 (s), 848 (m), 831 (m), 785 (m), 741 (s), 718 (m), 688 (s), 679 (m), 601 (s), 582 (m), 522 (m), 495 (m). Anal. calcd for C17H14N2S: C, 73.35; H, 5.07; N, 10.06. Found: C, 73.05; H, 5.06; N, 10.02.
N)CH2Ph-2-OMe (L6).
This product was prepared as described above for L4, starting from pyrrole-2-carboxyaldehyde (0.250 g, 2.63 mmol) and 2-methoxybenzylamine (0.358 g, 2.63 mmol). L6 was recovered as pale brown needles (0.334 g, 60%). Mp: 62.2 °C. 1H NMR (300 MHz, CDCl3, 25 °C): δ 3.80 (s, 3H, CH3), 4.73 (s, 2H, CH2), 6.19 (t, 3J = 6.0 Hz, 1H, 4-pyrrole), 6.47 (d, 2J = 3.0 Hz, 1H, 3-pyrrole), 6.74 (s, 1H, 5-pyrrole), 6.87–6.92 (m, 2H, Ar–H), 7.21–7.24 (m, 2H, Ar–H), 8.14 (s, 1H, CHN). 13C NMR (125 MHz, CDCl3, 25 °C): δ 55.40 (CH3), 58.79 (CH2), 109.58 (CH, 4-pyrrole), 110.30 (CH, 3-pyrrole), 114.37 (CH, Ar–C), 120.64 (CH, 5-pyrrole), 122.00 (CH, Ar–C), 127.90 (C, Ar–C), 128.29 (CH, Ar–C), 129.41 (CH, Ar–C), 130.43 (C, 2-pyrrole), 153.04 (CH, NC–H), 157.22 (C, Ar–C). IR (KBr, cm−1) ν: 3424 (sh), 3170 (w), 3123 (w), 3063 (w), 2956 (w), 2832 (w), 1639 (s), 1601 (w), 1592 (w), 1490 (m), 1460 (m), 1441 (m), 1422 (m), 1353 (m), 1315 (w), 1283 (m), 1244 (s), 1166 (w), 1136 (m), 1110 (m), 1031 (s), 975 (w), 879 (w), 837 (w), 757 (s), 739 (s), 606 (w), 581 (w). Anal. calcd for C13H14N2O: C, 72.87; H, 6.59; N, 13.07. Found: C, 72.81; H, 6.68; N, 13.20.
N)Ph-2-OPh (L4).
To a stirred solution containing pyrrole-2-carboxyaldehyde (0.250 g, 2.63 mmol) in ethanol (75 mL), 2-phenoxyaniline (0.560 g, 2.63 mmol) was added. The reaction mixture was stirred for 72 h at 65 °C. The solvent was evaporated to give a pale brown solid that was recrystallized from ethanol/ether to give L4 as an off-white solid (0.413 g, 87%). Mp: 110.4 °C. 1H NMR (300 MHz, CDCl3, 25 °C): δ 6.21 (dd, 2J = 2.6 and 3.6 Hz, 1H, H-pyrrole), 6.60 (dd, 2J = 1.3 and 3.6 Hz, 1H, H-py), 6.79 (d, 2J = 1.3 Hz, 1H, H-pyrrole), 6.90–7.27 (m, 9H, Ar–H), 8.23 (s, 1H, CHN), 9.69 (br s, 1H, NH-pyrrole). 13C NMR (125 MHz, CDCl3, 25 °C): δ 110.38 (CH, 4-pyrrole), 116.68 (CH, 3-pyrrole), 117.89 (2CH, Ar–C), 120.98 (CH, 5-pyrrole), 121.20 (CH, Ar–C), 122.56 (CH, Ar–C), 123.31 (CH, Ar–C), 124.82 (CH, Ar–C), 126.25 (CH, Ar–C), 129.61 (2CH, Ar–C), 130.98 (C, 2-pyrrole), 144.04 (C, Ar–C), 149.21 (C, Ar–C), 150.88 (CH, NC–H), 158.27 (C, Ar–C). IR (KBr, cm−1) ν: 3238 (s), 3095 (w), 3063 (w), 2979 (w), 2896 (w), 1687 (m), 1631 (s), 1594 (m), 1488 (s), 1455 (m), 1418 (m), 1339 (w), 1319 (w), 1267 (w), 1237 (s), 1205 (m), 1184 (w), 1159 (s), 1134 (m), 1090 (m), 1069 (w), 969 (w), 940 (w), 878 (m), 845 (m), 795 (m), 751 (s), 693 (m), 603 (m), 490 (w), 460 (w). Anal. calcd for C17H14N2O: C, 77.84; H, 5.38; N, 10.68. Found: C, 77.26; H, 5.09; N, 10.69.
N)CH2Ph-2-SPh (L5).
This product was prepared as described above for L4, starting from pyrrole-2-carboxyaldehyde (0.250 g, 2.63 mmol) and 2-(phenylthio)aniline (0.529 g, 2.63 mmol). L5 was recovered as an off-white solid (0.219 g, 30%). Mp: 102.5 °C. 1H NMR (300 MHz, CDCl3, 25 °C): δ 6.30 (dd, 2J = 2.7 and 3.5 Hz, 1H, H-pyrrole), 6.67 (dd, 2J = 1.1 and 3.6 Hz, 1H, H-pyrrole), 6.96–7.07 (m, 4H, H–Ar + H-pyrrole), 7.16–7.22 (m, 1H, H–Ar), 7.30–7.37 (m, 3H, H–Ar), 7.44–7.47 (m, 2H, H–Ar), 8.16 (s, 1H, CHN), 9.55 (br s, 1H, NH-pyrrole). 13C NMR (125 MHz, CDCl3, 25 °C): δ 110.55 (CH, 4-pyrrole), 116.70 (CH, 3-pyrrole), 118.42 (CH, 5-pyrrole), 123.38 (CH, Ar–C), 125.88 (CH, Ar–C), 127.14 (CH, Ar–C), 127.84 (CH, Ar–C), 128.93 (2CH, Ar–C), 129.37 (2CH, Ar–C), 130.93 (CH, Ar–C), 132.46 (C, 2-pyrrole), 133.46 (C, Ar–C), 134.19 (C, Ar–C), 149.77 (CH, NC–H), 149.87 (C, Ar–C). IR (KBr, cm−1) ν: 3215 (m), 3165 (w), 3053 (w), 2975 (w), 2906 (w), 2864 (w), 1615 (s), 1562 (s), 1551 (m), 1463 (m), 1438 (m), 1405 (s), 1339 (m), 1315 (m), 1302 (w), 1262 (m), 1242 (w), 1198 (s), 1131 (s), 1088 (s), 1058 (m), 1032 (s), 962 (w), 935 (w), 912 (w), 879 (s), 848 (m), 831 (m), 785 (m), 741 (s), 718 (m), 688 (s), 679 (m), 601 (s), 582 (m), 522 (m), 495 (m). Anal. calcd for C17H14N2S: C, 73.35; H, 5.07; N, 10.06. Found: C, 73.05; H, 5.06; N, 10.02.
N)CH2Ph-2-OMe (L6).
This product was prepared as described above for L4, starting from pyrrole-2-carboxyaldehyde (0.250 g, 2.63 mmol) and 2-methoxybenzylamine (0.358 g, 2.63 mmol). L6 was recovered as pale brown needles (0.334 g, 60%). Mp: 62.2 °C. 1H NMR (300 MHz, CDCl3, 25 °C): δ 3.80 (s, 3H, CH3), 4.73 (s, 2H, CH2), 6.19 (t, 3J = 6.0 Hz, 1H, 4-pyrrole), 6.47 (d, 2J = 3.0 Hz, 1H, 3-pyrrole), 6.74 (s, 1H, 5-pyrrole), 6.87–6.92 (m, 2H, Ar–H), 7.21–7.24 (m, 2H, Ar–H), 8.14 (s, 1H, CHN). 13C NMR (125 MHz, CDCl3, 25 °C): δ 55.40 (CH3), 58.79 (CH2), 109.58 (CH, 4-pyrrole), 110.30 (CH, 3-pyrrole), 114.37 (CH, Ar–C), 120.64 (CH, 5-pyrrole), 122.00 (CH, Ar–C), 127.90 (C, Ar–C), 128.29 (CH, Ar–C), 129.41 (CH, Ar–C), 130.43 (C, 2-pyrrole), 153.04 (CH, NC–H), 157.22 (C, Ar–C). IR (KBr, cm−1) ν: 3424 (sh), 3170 (w), 3123 (w), 3063 (w), 2956 (w), 2832 (w), 1639 (s), 1601 (w), 1592 (w), 1490 (m), 1460 (m), 1441 (m), 1422 (m), 1353 (m), 1315 (w), 1283 (m), 1244 (s), 1166 (w), 1136 (m), 1110 (m), 1031 (s), 975 (w), 879 (w), 837 (w), 757 (s), 739 (s), 606 (w), 581 (w). Anal. calcd for C13H14N2O: C, 72.87; H, 6.59; N, 13.07. Found: C, 72.81; H, 6.68; N, 13.20.
Synthesis of the Ni(II) complexes
N)C2H4(NH)Ph}Cl] (Ni1).
To a solution of L1 (0.200 g, 0.94 mmol) in THF (5 mL) was added dropwise a suspension of NaH (0.034 g, 0.94 mmol) in THF (5 mL) at 0 °C. The resulting red solution was stirred for 4 h at room temperature and then added dropwise to a solution of NiCl2(dme) (0.205 g, 0.94 mmol) in THF (10 mL) at −78 °C. The reaction mixture was stirred overnight at room temperature and then volatiles were removed in vacuo. The brown crude solid was dissolved in toluene (15 mL), filtered by cannula and the resulting solution was concentrated (ca. 3 mL). Then, pentane (15 mL) was added to afford a pale brown solid which was washed with diethyl ether (2 × 10 mL) to give, after drying, Ni1 as a brown solid (0.210 g, 73%). IR (ATR, cm−1): ν 3048 (w), 2954 (w), 2845 (w), 1626 (w), 1597 (w), 1576 (s), 1490 (m), 1441 (m), 1387 (m), 1301 (s), 1246 (w), 1030 (s), 733 (s), 690 (s), 601 (m), 508 (m). Anal. calcd for C13H14ClNiN3: C, 50.96; H, 4.61; N, 13.71. Found: C, 51.27; H, 4.94; N, 13.60.
N)C2H4(NH)Ph}Cl] (Ni2).
This product was prepared as described above for Ni1, starting from L2 (0.183 g, 0.68 mmol), NaH (0.025 g, 0.68 mmol), and NiCl2(dme) (0.150 g, 0.68 mmol) in THF (10 mL) to give Ni2 as a pale brown solid (0.221 g, 90%). IR (ATR, cm−1): ν 3190 (w), 3102 (w), 2959 (w), 2899 (w), 1639 (m), 1593 (s), 1491 (m), 1441 (m), 1392 (m), 1341 (m), 1269 (m), 1231 (m), 1153 (w), 1093 (w), 1044 (s), 914 (m), 785 (m), 750 (s), 690 (s), 511 (m). Anal. calcd for C17H22ClNiN3: C, 56.32; H, 6.12; N, 11.59. Found: C, 56.11; H, 5.87; N, 11.44.
N)C2H4OPh}Cl] (Ni3).
This product was prepared as described above for Ni1, starting from L3 (0.199 g, 0.93 mmol), NaH (0.034 g, 0.93 mmol), and NiCl2(dme) (0.190 g, 0.93 mmol) in THF (10 mL) to give Ni3 as a pale brown solid (0.193 g, 67%). IR (ATR, cm−1): ν 3011 (w), 2933 (w), 2924 (w), 2861 (w), 1586 (s), 1487 (m), 1487 (m), 1443 (w), 1391 (m), 1312 (m), 1231 (s), 1198 (w), 1172 (m), 1105 (w), 1079 (w), 1050 (m), 1034 (s), 1003 (w), 962 (w), 936 (w), 891 (w), 821 (w), 800 (w), 782 (w), 758 (s), 722 (s), 689 (w), 673 (m), 600 (m), 548 (w), 509 (m), 486 (w), 450 (s). Anal. calcd for C13H13ClNiN2O: C, 50.79; H, 4.26; N, 9.11. Found: C, 51.15; H, 4.46; N, 8.88.
N)Ph-2-OPh}Cl] (Ni4).
This product was prepared as described above for Ni1, starting from L4 (0.299 g, 1.14 mmol), NaH (0.042 g, 1.14 mmol), and NiCl2(dme) (0.250 g, 1.14 mmol) in THF (10 mL) to give Ni4 as a pale brown solid (0.280 g, 69%). IR (ATR, cm−1): ν 3234 (w), 3065 (w), 2976 (w), 2899 (w), 1626 (s), 1573 (m), 1483 (s), 1452 (m), 1415 (s), 1332 (w), 1312 (w), 1268 (w), 1232 (s), 1182 (s), 1090 (s), 1036 (s), 968 (w), 874 (m), 835 (m), 795 (m), 746 (s), 690 (s), 601 (m), 490 (m). Anal. calcd for C17H13ClNiN2O: C, 57.44; H, 3.69; N, 7.88. Found: C, 57.05; H, 3.44; N, 7.66.
N)CH2Ph-2-SPh}Cl] (Ni5).
This product was prepared as described above for Ni1, starting from L5 (0.326 g, 1.17 mmol), NaH (0.043 g, 1.17 mmol), and NiCl2(dme) (0.255 g, 1.17 mmol) in THF (10 mL) to give Ni5 as a pale brown solid (0.377 g, 87%). IR (ATR, cm−1): ν 3054 (w), 1590 (m), 1556 (s), 1497 (s), 1474 (m), 1463 (m), 1437 (m), 1379 (m), 1291 (s), 1225 (w), 1185 (m), 1164 (w), 1074 (w), 1030 (s), 996 (w), 943 (w), 902 (m), 889 (w), 836 (w), 748 (s), 723 (m), 685 (m), 667 (w), 625 (w), 597 (m). Anal. calcd for C17H13ClNiN2S: C, 54.96; H, 3.53; N, 7.54. Found: C, 54.81; H, 3.94; N, 7.09.
N)CH2Ph-2-OMe}Cl] (Ni6).
This product was prepared as described above for Ni1, starting from L6 (0.300 g, 1.40 mmol), NaH (0.051 g, 1.40 mmol), and NiCl2(dme) (0.307 g, 1.40 mmol) in THF (10 mL) to give Ni6 as a pale brown solid (0.271 g, 63%). IR (ATR, cm−1): ν 2999 (w), 2924 (w), 2835 (w), 1618 (w), 1600 (m), 1579 (s), 1490 (m), 1435 (m), 1378 (m), 1291 (m), 1241 (s), 1175 (w), 116 (m), 1026 (s), 805 (w), 730 (s), 601 (w). Anal. calcd for C13H13ClNiN2O: C, 50.79; H, 4.26; N, 9.11. Found: C, 51.34; H, 4.96; N, 8.97.
General oligomerization procedure
Ethylene oligomerization reactions were performed in a 100 mL double-walled stainless Parr reactor equipped with mechanical stirring, internal temperature control and continuous feed of ethylene. The Parr reactor was dried in an oven at 120 °C for 5 h prior to each run, and then cooled under vacuum for 30 min. A typical reaction was performed by introducing toluene (30 mL) and the proper amount of co-catalyst (MAO or EASC) into the reactor under an ethylene atmosphere. After 20 min, the toluene catalyst solution (10 mL, [Ni] = 10 μmol) was injected into the reactor under a stream of ethylene and then the reactor was immediately pressurized. Ethylene was continuously fed in order to maintain the desired ethylene pressure. After 20 min, the reaction was stopped by cooling the system to −60 °C and depressurizing. An exact amount of cyclohexane was introduced (as an internal standard) and the mixture was analyzed by quantitative GLC.X-ray diffraction analyses
A suitable single-crystal of Ni1 was mounted onto a glass fiber using the ‘‘oil-drop’’ method. Diffraction data were collected at 150(2) K using an APEXII Bruker-AXS diffractometer with graphite-monochromatized MoKα radiation (λ = 0.71073 Å). A combination of ω and φ scans was carried out to obtain at least a unique data set. The crystal structures were solved by direct methods, remaining atoms were located from difference Fourier synthesis followed by full-matrix least-squares refinement based on F2 (programs SIR97 and SHELXL-97) with the aid of the WINGX program. All non-hydrogen atoms were refined with anisotropic atomic displacement parameters. H atoms were finally included in their calculated positions.Acknowledgements
This work was supported in part by the Petrobras S/A, CAPES, French MESR, and CNRS. The authors are grateful to CAPES-COFECUB for joined Action 804/14 and CAPES-CNRS for joined action PICS05923.Notes and references
- (a) M. Peuckert and W. Keim, Organometallics, 1983, 2, 594 CrossRef CAS; (b) M. Peuckert, W. Keim, S. Storp and R. S. Weber, J. Mol. Catal. A: Chem., 1983, 20, 115 CrossRef CAS; (c) W. Keim, Angew. Chem., Int. Ed., 1990, 29, 235 CrossRef PubMed; (d) S. M. Pillai, M. Ravindranathan and S. Sivaram, Chem. Rev., 1986, 86, 353 CrossRef CAS; (e) J. Skupinska, Chem. Rev., 1991, 91, 613 CrossRef CAS; (f) P.-A. R. Breuil, L. Magna and H. Olivier-Bourbigou, Catal. Lett., 2015, 145, 173–192 CrossRef CAS.
- (a) D. Vogt, in Applied Homogeneous Catalysis with Organometallic Compounds, ed. B. Cornils and W. A. Herrmann, Wiley-VCH, Inc., Weinheim, 2000, pp. 245–258 Search PubMed; (b) P. W. N. N. van Leeuwen, Homogeneous Catalysis, Kluwer Academic, Inc., Dordrecht, 2004, pp. 175–190 Search PubMed.
- (a) J. N. L. Dennett, A. L. Gillon, K. Heslop, D. J. Hyett, J. S. Fleming, C. E. Lloyd-Jones, A. G. Orpen, P. G. Pringle and D. F. Wass, Organometallics, 2004, 23, 6077 CrossRef CAS; (b) C. Bianchini, L. Gonsalvi, W. Oberhauser, D. Sémeril, P. Brüggeller and R. J. Gutmann, J. Chem. Soc., Dalton Trans., 2003, 3869–3875 RSC; (c) T. M. J. Anselment, S. I. Vagin and B. Rieger, Dalton Trans., 2008, 4537–4548 RSC; (d) Z. Guan and W. J. Marshall, Organometallics, 2002, 21, 3580–3586 CrossRef CAS; (e) P. Boulens, M. Lutz, E. Jeanneau, H. Olivier-Bourbigou, J. N. H. Reek and P.-A. R. Breuil, Eur. J. Inorg. Chem., 2014, 3754–3762 CrossRef CAS PubMed; (f) A. Ghisolfi, C. Fliedel, V. Rosa, K. Y. Monakhov and P. Braunstein, Organometallics, 2014, 33, 2523–2534 CrossRef CAS.
- (a) F. Speiser, P. Braunstein, L. Saussine and R. Welter, Organometallics, 2004, 23, 2613–2624 CrossRef CAS; (b) F. Speiser, P. Braunstein and L. Saussine, Organometallics, 2004, 23, 2625–2632 CrossRef CAS; (c) F. Speiser, P. Braunstein and L. Saussine, Organometallics, 2004, 23, 2633–2640 CrossRef CAS; (d) H. P. Chen, Y. H. Liu, S. M. Peng and S. T. Liu, Organometallics, 2003, 22, 4893–4899 CrossRef CAS; (e) E. K. van den Beuken, W. J. J. Smeets, A. L. Spek and B. L. Feringa, Chem. Commun., 1998, 223–224 RSC; (f) P. Braunstein, J. Pietsch, Y. Chauvin, S. Mercier, L. Saussine, A. DeCian and J. Fischer, J. Chem. Soc., Dalton Trans., 1996, 3571–3574 RSC; (g) J. Pietsch, P. Braunstein and Y. Chauvin, New J. Chem., 1998, 467–472 RSC; (h) F. Speiser, P. Braunstein, L. Saussine and R. Welter, Inorg. Chem., 2004, 43, 1649–1658 CrossRef CAS PubMed; (i) X. Tang, D. Zhang, S. Jie, W. H. Sun and J. Chen, J. Organomet. Chem., 2005, 690, 3918–3928 CrossRef CAS PubMed; (j) F. Speiser, P. Braunstein and L. Saussine, Acc. Chem. Res., 2005, 38, 784–793 CrossRef CAS PubMed; (k) W.-H. Sun, W. Zhang, T. Gao, X. Tang, L. Chen, Y. Li and X. Jin, J. Organomet. Chem., 2004, 689, 917–929 CrossRef CAS PubMed; (l) M. E. Bluhm, C. Folli, O. Walter and M. Döring, J. Mol. Catal. A: Chem., 2005, 229, 177–181 CrossRef CAS PubMed; (m) Z. Weng, S. Teo and T. S. A. Hor, Organometallics, 2006, 25, 4878–4882 CrossRef CAS; (n) L. O. de la Tabla, I. Matas, P. Palma, E. Álvarez and J. Cámpora, Organometallics, 2012, 31, 1006–1016 CrossRef; (o) S. Zhang, R. Pattacini, S. Jie and P. Braunstein, Dalton Trans., 2012, 41, 379 RSC.
- (a) W. Keim, Angew. Chem., Int. Ed., 1990, 29, 235–244 CrossRef PubMed; (b) J. M. Malinoski and M. Brookhart, Organometallics, 2003, 22, 5324–5335 CrossRef CAS; (c) W. Liu, J. M. Malinoski and M. Brookhart, Organometallics, 2002, 21, 2836–2838 CrossRef CAS; (d) J. Heinicke, M. Köhler, N. Peulecke and W. Keim, J. Catal., 2004, 225, 16–23 CrossRef CAS PubMed; (e) P. Kuhn, D. Sémeril, D. Matt, M. J. Chetcutti and P. Lutz, Dalton Trans., 2007, 515–528 RSC; (f) A. Kermagoret and P. Braunstein, Dalton Trans., 2008, 822–831 RSC; (g) C.-Y. Guo, N. Peulecke, M. K. Kindermann and J. W. Heinicke, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 258–266 CrossRef CAS PubMed; (h) J. Flapper, H. Kooijman, M. Lutz, A. L. Spek, P. W. N. M. van Leeuwen, C. J. Elsevier and P. C. J. Kamer, Organometallics, 2009, 28, 1180–1192 CrossRef CAS.
- (a) H. Liu, W. Zhao, X. Hao, C. Redshaw, W. Huang and W.-H. Sun, Organometallics, 2011, 30, 2418–2424 CrossRef CAS; (b) R. Gao, L. Xiao, X. Hao, W.-H. Sun and F. Wang, Dalton Trans., 2008, 5645–5651 RSC; (c) X. Tang, W.-H. Sun, T. Gao, J. Hou, J. Chen and W. Chen, J. Organomet. Chem., 2005, 690, 1570–1580 CrossRef CAS PubMed; (d) S. Jie, D. Zhang, T. Zhang, W.-H. Sun, J. Chen, Q. Ren, D. Liu, G. Zheng and W. Chen, J. Organomet. Chem., 2005, 690, 1739–1749 CrossRef CAS PubMed; (e) E. Nelkenbaum, M. Kapon and M. S. Eisen, J. Organomet. Chem., 2005, 690, 2297–2305 CrossRef CAS PubMed; (f) J. M. Benito, E. de Jesús, F. J. de la Mata, J. C. Flores, R. Gómez and P. Gómez-Sal, Organometallics, 2006, 25, 3876–3887 CrossRef CAS; (g) C.-L. Song, L.-M. Tang, Y.-G. Li, X.-F. Li, J. Chen and Y.-S. Li, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 1964–1974 CrossRef CAS PubMed; (h) B. Y. Lee, X. Bu and G. C. Bazan, Organometallics, 2001, 20, 5425–5431 CrossRef CAS; (i) S. A. Sveida and M. Brookhart, Organometallics, 1999, 18, 65–74 CrossRef; (j) C. M. Killian, L. K. Johnson and M. Brookhart, Organometallics, 1997, 16, 2005–2007 CrossRef CAS; (k) A. J. Swarts and S. F. Mapolie, Dalton Trans., 2014, 43, 9892–9900 RSC; (l) J. Canivet, S. Aguado, Y. Schuurman and D. Farrusseng, J. Am. Chem. Soc., 2013, 135, 4195–4198 CrossRef CAS PubMed; (m) S. Song, T. Xiao, T. Liang, F. Wang, C. Redshaw and W.-H. Sun, Catal. Sci. Technol., 2011, 1, 69–75 RSC; (n) J. Yu, X. Hu, Y. Zeng, L. Zhang, C. Ni, X. Haoc and W.-H. Sun, New J. Chem., 2011, 35, 178–183 RSC; (o) R. M. Bellabarba, P. T. Gomes and S. I. Pascu, Dalton Trans., 2003, 4431–4436 RSC; (p) J. Li, T. Gao, W. Zhang and W.-H. Sun, Inorg. Chem. Commun., 2003, 6, 1372–1374 CrossRef CAS PubMed.
- (a) T. R. Younkin, E. F. Connor, J. I. Henderson, S. K. Friedrich, R. H. Grubbs and D. A. Bansleben, Science, 2000, 287, 460–462 CrossRef CAS; (b) C. Carlini, M. Isola, V. Liuzzo, A. M. R. Galletti and C. Sbrana, Appl. Catal., A, 2002, 231, 307–320 CrossRef CAS; (c) S. Wu and S. Lu, Appl. Catal., A, 2003, 246, 295–301 CrossRef CAS; (d) F. Speiser, P. Braunstein and L. Saussine, Inorg. Chem., 2004, 43, 4234–4240 CrossRef CAS PubMed; (e) Y. Chen, G. Wu and G. C. Bazan, Angew. Chem., Int. Ed., 2005, 44, 1108–1112 CrossRef CAS PubMed; (f) A. Kermagoret and P. Braunstein, Dalton Trans., 2008, 1564–1573 RSC; (g) N. A. Cooley, S. M. Green and D. F. Wass, Organometallics, 2001, 20, 4769–4771 CrossRef CAS; (h) T. Hu, L.-M. Tang, X.-F. Li, Y.-S. Li and N.-H. Hu, Organometallics, 2005, 24, 2628–2632 CrossRef CAS; (i) T. Cheisson, T.-P.-A. Cao, X. F. le Goff and A. Auffrant, Organometallics, 2014, 33, 6193–6199 CrossRef CAS.
- (a) L. Xiao, M. Zhang, R. Gao, X. Cao and W.-H. Sun, Aust. J. Chem., 2010, 63, 109 CrossRef CAS; (b) Q.-Z. Yang, A. Kermagoret, M. Agostinho, O. Siri and P. Braunstein, Organometallics, 2006, 25, 5518 CrossRef CAS; (c) A. Kermagoret, F. Tomicki and P. Braunstein, Dalton Trans., 2008, 2945 RSC; (d) J. Hou, W.-H. Sun, S. Zhang, H. Ma, Y. Deng and X. Lu, Organometallics, 2006, 25, 236 CrossRef CAS; (e) C. Zhang, W.-H. Sun and Z.-X. Wang, Eur. J. Inorg. Chem., 2006, 4895 CrossRef CAS PubMed; (f) W.-H. Sun, S. Zhang, S. Jie, W. Zhang, Y. Li, H. Ma, J. Chen, K. Wedeking and R. Fröhlich, J. Organomet. Chem., 2006, 691, 4196 CrossRef CAS PubMed; (g) W.-H. Sun, K. Wang, K. Wedeking, D. Zhang, S. Zhang, J. Cai and Y. Li, Organometallics, 2007, 26, 4781 CrossRef CAS; (h) P. Hao, S. Zhang, W.-H. Sun, Q. Shi, S. Adewuyi, X. Lu and P. Li, Organometallics, 2007, 26, 2439 CrossRef CAS; (i) Y. Chen, P. Hao, W. Zuo, K. Gao and W.-H. Sun, J. Organomet. Chem., 2008, 693, 1829 CrossRef CAS PubMed; (j) S. Adewuyi, G. Li, S. Zhang, W. Wang, P. Hao, W.-H. Sun, N. Tang and J. Yi, J. Organomet. Chem., 2007, 692, 3532 CrossRef CAS PubMed; (k) A. P. Armitage, Y. D. M. Champouret, H. Grigoli, J. D. A. Pelletier, K. Singh and G. A. Solan, Eur. J. Inorg. Chem., 2008, 4597 CrossRef CAS PubMed; (l) Y. Yang, P. Yang, C. Zhang, G. Li, X.-J. Yang, B. Wu and C. Janiak, J. Mol. Catal. A: Chem., 2008, 296, 9 CrossRef CAS PubMed; (m) R. Gao, M. Zhang, T. Liang, F. Wang and W.-H. Sun, Organometallics, 2008, 27, 5641 CrossRef CAS; (n) S. Jie, S. Zhang and W.-H. Sun, Eur. J. Inorg. Chem., 2007, 5584 CrossRef CAS PubMed; (o) M. Zhang, P. Hao, W. Zuo, S. Jie and W.-H. Sun, J. Organomet. Chem., 2008, 693, 483 CrossRef CAS PubMed; (p) W.-H. Sun, P. Hao, S. Zhang, Q. Shi, W. Zuo, X. Tang and X. Lu, Organometallics, 2007, 26, 2720 CrossRef CAS; (q) R. Gao, Y. Li, F. Wang, W.-H. Sun and M. Bochmann, Eur. J. Inorg. Chem., 2009, 4149 CrossRef CAS PubMed; (r) G. S. Nyamato, M. G. Alam, S. O. Ojwach and M. P. Akerman, J. Organomet. Chem., 2015, 783, 64–72 CrossRef CAS PubMed; (s) J. Lai, X. Hou, Y. Liu, C. Redshaw and W.-H. Sun, J. Organomet. Chem., 2012, 702, 52–58 CrossRef CAS PubMed; (t) C. Obuaha, B. Omondia, K. Nozaki and J. Darkwa, J. Mol. Catal. A: Chem., 2014, 382, 31–40 CrossRef PubMed.
- F. Speiser, P. Braunstein and L. Saussine, Dalton Trans., 2004, 1539 RSC.
- M. Zhang, S. Zhang, P. Hao, S. Jie, W.-H. Sun, P. Li and X. Lu, Eur. J. Inorg. Chem., 2007, 3816 CrossRef CAS PubMed.
- A. Boudier, P.-A. R. Breuil, L. Magna, H. Olivier-Bourbigou and P. Braunstein, J. Organomet. Chem., 2012, 718, 31–37 CrossRef CAS PubMed.
- (a) N. Ajellal, M. C. A. Kuhn, A. D. G. Boff, M. Hoerner, C. M. Thomas, J.-F. Carpentier and O. L. Casagrande, Organometallics, 2006, 25, 1213 CrossRef CAS; (b) L. L. de Oliveira, R. R. Campedelli, A. L. Bergamo, A. H. D. P. dos Santos and O. L. Casagrande, J. Braz. Chem. Soc., 2010, 21, 1318 CrossRef CAS; (c) F. A. Kunrath, R. F. de Souza, O. L. Casagrande, N. R. Brooks and V. G. Young, Organometallics, 2003, 22, 4739 CrossRef CAS; (d) F. Junges, M. C. A. Kuhn, A. H. P. Santos, C. R. K. Rabello, C. M. Thomas, J.-F. Carpentier and O. L. Casagrande Jr, Organometallics, 2007, 26, 4010 CrossRef CAS; (e) L. L. de Oliveira, R. R. Campedelli, M. C. A. Kuhn, J.-F. Carpentier and O. L. Casagrande Jr, J. Mol. Catal. A: Chem., 2008, 288, 58 CrossRef CAS PubMed; (f) A. H. P. S. Ulbrich, A. L. Bergamo and O. L. Casagrande Jr, Catal. Commun., 2011, 16, 245 CrossRef CAS PubMed; (g) E. Kirillov, T. Roisnel, A. Razavi and J.-F. Carpentier, Organometallics, 2009, 28, 2401 CrossRef CAS.
- J. Wang, L. Wan, D. Zhang, Q. Wang and Z. Chen, Eur. J. Inorg. Chem., 2013, 2093 CrossRef CAS PubMed.
- (a) X. Yang and Z.-X. Wang, Organometallics, 2014, 33, 5863 CrossRef CAS; (b) F.-B. Han, Y.-L. Zhang, X.-L. Sun, B.-G. Li, Y.-H. Guo and Y. Tang, Organometallics, 2008, 27, 1924 CrossRef CAS; (c) L.-C. Liang, P.-S. Chien, J.-M. Lin, M.-H. Huang, Y.-L. Huang and J.-H. Liao, Organometallics, 2006, 25, 1399 CrossRef CAS.
- S. Wang, W.-H. Sun and C. Redshaw, J. Organomet. Chem., 2014, 751, 717–741 CrossRef CAS PubMed.
- A. C. Pinheiro, E. Kirillov, T. Roisnel, J.-F. Carpentier and O. L. Casagrande Jr, Dalton Trans., 2015, 44, 16073 RSC.
- H. J. Anderson and C. E. Loader, Synthesis, 1985, 353 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data for Ni1, and typical GLC analyses of oligomerization mixtures. CCDC 1407832. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra16782e |
| This journal is © The Royal Society of Chemistry 2015 |