DOI:
10.1039/C5RA16730B
(Paper)
RSC Adv., 2015,
5, 88181-88190
Novel polyamides with fluorene-based triphenylamine: electrofluorescence and electrochromic properties†
Received
10th September 2015
, Accepted 12th October 2015
First published on 12th October 2015
Abstract
A series of novel polyamides with fluorene-based triphenylamine units in the backbone were prepared from a newly synthesized diamine monomer, N,N-di (4-aminophenyl)-2-amino-9,9-dimethylfluorene, and various dicarboxylic acids via phosphorylation polyamidation technique. These polyamides were amorphous and exhibited excellent solubility in many polar solvents and could be solution-cast into flexible polymer films. They possess good thermal stability with glass transition temperature in the range of 256–316 °C and 10% weight loss in excess of 500 °C in nitrogen. The dilute N-methyl-2-pyrrolidone (NMP) solutions of these polyamides showed strong UV-vis absorption bands at 334–367 nm and the polyamide derived from 1,4-cyclohexanedicarboxylic acid exhibited fluorescence maximum at 441 nm with quantum yields up to 47.1%. Furthermore, the fluorescence can be effectively switched between “on” and “off” states by applying reduction and oxidation potentials, exhibiting a high contrast ratio (If/If0) of 12.7. The polymer films showed reversible electrochemical oxidation and reduction, enabling an improved stability of electrochromic characteristics with color changing from colorless to green. The anodically electrochromic films had high coloration efficiency (up to 283 cm2 C−1 at 860 nm) and after over 500 cycles the polymer films still exhibited continuous cyclic stability of electrochromism.
Introduction
Electrochromism refers to the reversible changes in color during reduction or oxidation. Since its discovery in inorganic materials,1 these properties had aroused great interest of scientists over the past decades.2 Except for inorganic materials, many conducting polymers such as polyanilines, polypyrroles, and polythiophenes, also play important roles in electrochromic field.3 Moreover, the conducting polymers have several advantages over inorganic compounds as electrochromic materials, including fast switching time, outstanding coloration efficiency and the possibility in fabricating large-area devices.4 In recent years, triphenylamine-containing condensation-type polymers have been developed as a new and attractive family of electrochromic materials because of the colorless neutral state.5 Electrochromic properties have been proved useful in the rear-view mirrors,6 electronic paper,7 energy saving smart window,8 and high contrast displays.9 Yet to see them to meet the real-life applications, electrochromic materials need to exhibit long-term stability, rapid redox switching and distinguishing transmittance change between their bleached and colored states.10 The difficulties in achieving satisfactory values for all these parameters at the same time stimulate the development of new methods of preparation of electrochromic films, new materials and components for the devices.11
Fluorene is a biphenyl structure with rigid plane and large π-conjugated system, whose derivatives have been widely used in organic electro-luminescence materials,12 two-photon absorbing materials,13 photochromic materials14 and solar cells15 because of their extremely high photoluminescence (PL), electroluminescence quantum efficiencies, thermal stability, good solubility and facile functionalization at the C-9 position. In particular, fluorene-based polymers are regarded as one of the most potential candidates for efficient blue luminescence materials in OLED (Organic Light-Emitting Diode) yield.16 In recent years, a vast number of fluorene derivatives have been reported due to the attractive properties as mentioned above. Among these derivatives, fluorene-triphenylamine system including tetratriphenylamine-fluorene derivatives, star-shaped with triphenylamine-cored and fluorene-arms derivatives, polyfluorene with triphenylamine as end group, side or main segment have been found to exhibit highly enhanced hole-transporting properties and increased emitting efficiency of the devices, thus, they can be used as hole-transporting emitters in the OLEDs.17 Moreover, by using the “electron-transfer” fluorescence quenching mechanisms, the luminescence of the fluorene-triphenylamine derivatives could be reversibly switched by changing the redox states of the triphenylamine units,18 which will greatly expand the applications of these derivatives.
Triphenylamine derivatives are well known for their electroactive and photoactive properties, which make them useful as photoconductors, hole-transporters, light-emitters, memory devices and electrochromic materials.19 Electron-rich triphenylamine can be easily oxidized to form stable radical cations. However, according to some reports before, unsubstituted triphenylamine is not stable, undergoing coupling deprotonation to form tetraphenylbenzidine after the formation of the initial monocation radical.20 To prevent the irreversible coupling reactions, appropriate substituents (such as methoxy and methyl) could be introduced at the para-position of the triphenylamine unit, which afford stable radical cations.5b,21 Different from the former study, our strategy was to design new structure to protect the para-position of triphenylamine. To our knowledge, there has been no report on fluorene-based triphenylamine condensation polymers. In this report, considering these attractive properties associated with the fluorene and triphenylamine units, we synthesize a new diamine monomer and its derived polyamides.
Introducing fluorene-based triphenylamine unit into the polymer backbone will not only improve the solubility of the polymers and prevent coupling reactions by protecting the para position of triphenylamine, but also lead to intriguing electroswitchable-fluorescence and electrochromic properties, which is of great value for optoelectronic applications.
Experiment
Materials
4,4′-Diaminotriphenylamine was synthesized according to a previously reported procedure.22 2-Amino-9,9-dimethylfluorene (TCI), 4-fluoronitrobenzene (Acros), 10% palladium on charcoal (Pd/C, TCI), hydrazine monohydrate (TCI), cesium fluoride (CsF, Acros), triphenyl phosphite (TPP, Acros) and dimethyl sulfoxide (DMSO) were used as received. Pyridine and NMP were purified by vacuum distillation over CaH2 and stored over 4 Å molecular sieves prior to use. Commercially available dicarboxylic acids that include terephthalic acid (3a, TCI), 4,4′-biphenyldicarboxylic acid (3b, TCI), 4,4′-dicarboxydiphenylether (3c, TCI), 2,2-bis(4-carboxy-phenyl)hexafluoropropane (3d, TCI), 1,4-cyclohexane-dicarboxylic acid (3e, TCI) were used as received. Calcium chloride was dried under vacuum at 180 °C for 12 h prior to use. Tetrabutylammonium perchlorate (TBAP, Acros) was recrystallized twice from ethanol under a nitrogen atmosphere and then dried in vacuo before use. The other commercially available reagents and solvents were used without further purification.
Synthesis of monomers
Synthesis of N,N-di(4-nitrophenyl)-2-amino-9,9-dimethylfluorene. In a 250 mL three-neck round-bottom flask equipped with a stirring bar, a mixture of 12.0 g (57 mmol) of 2-amino-9,9-dimethyl-fluorene, 16.9 g (120 mmol) of 4-fluoronitrobenzene and 18.2 g (120 mmol) of CsF in 100 mL of dried DMSO was heated with stirring at 150 °C for 12 h under nitrogen atmosphere. After cooling down to 30 °C, the mixture was poured into 500 mL stirred ethanol slowly and the yellow precipitate was collected by filtration and washed thoroughly by ethanol and hot water. The crude product was filtered and recrystallized from glacial acetic acid to afford 21.2 g in 80% yield; mp = 195 °C; IR (KBr): 1308 cm−1, 1577 cm−1 (–NO2 stretch). 1H NMR (300 MHz, DMSO-d6, δ, ppm): 8.21 (d, J = 3.3 Hz, 4H), 7.95 (d, J = 6.6 Hz, 1H), 7.87–7.84 (m, 1H), 7.58–7.55 (m, 1H), 7.51 (d, J = 1.8 Hz, 1H), 7.4–7.3 (m, 2H), 7.26 (d, J = 9.3 Hz, 4H), 7.24 (m, 1H), 1.42 (s, 6H, –CH3).
Synthesis of N,N-di(4-aminophenyl)-2-amino-9,9-dimethylfluorene. In a 500 mL three-neck round-bottomed flask equipped with a stirring bar under nitrogen atmosphere, 20.0 g (44 mmol) of dinitro compound and 2.0 g 10% Pd/C were dissolved in 200 mL ethanol. The suspension solution was heated to reflux, and 50 mL of hydrazine monohydrate was added slowly to the mixture, then the solution was stirred at reflux temperature. After a further 12 h of reflux, the solution was filtered to remove Pd/C, and the filtrate was cooled under a nitrogen flow to grow green crystals. The crystals were collected by filtration and dried in vacuo at 80 °C to give 14.2 g (80% in yield) with a mp of 240 °C; IR (KBr): 3445 cm−1, 3362 cm−1 (NH2 stretch). 1H NMR (300 MHz, DMSO-d6, δ, ppm): 7.57 (d, J = 7.6 Hz, 1H, Hf), 7.48 (d, J = 8.4 Hz, 1H, Hd), 7.41 (d, J = 7.4 Hz, 1H, Hi), 7.23 (t, J = 7.3 Hz, 1H, Hg), 7.14 (t, J = 7.3 Hz, 1H, Hh), 6.84 (d, J = 8.3 Hz, 4H, Hb), 6.75 (s, 1H, He), 6.59–6.53 (m, 5H, Hc + Ha), 5.0 (s, 4H, NH2), 1.28 (s, 6H, CH3). 13C HMR (75 MHz, DMSO-d6, δ, ppm): 154.69 (C16), 152.98 (C14), 150.01 (C9), 145.91 (C1), 139.52 (C5), 136.69 (C4), 129.33 (C8), 127.66 (C3), 127.32 (C10), 125.83 (C11), 122.83 (C12), 120.83 (C17), 119.00 (C13), 116.58 (C7), 115.34 (C2), 111.39 (C6), 46.52 (C15), 27.55 (–CH3).
Synthesis of polyamides
The synthesis of polyamides 4c was used as an example to illustrate the general synthesizing route used to produce these polyamides. Into a 50 mL three-neck round-bottomed flask were added 0.3912 g (1 mmol) of diamine 2, 0.2582 g (1 mmol) of 4, 4′-dicarboxydiphenyl ether (3c), 0.15 g of dried calcium chloride, 1 mL of triphenyl phosphite, 0.5 mL of pyridine and 5 mL of NMP. The reaction mixture was stirred at 115 °C in nitrogen atmosphere for 3 h. After cooling, the obtained highly viscous polymer solution was poured slowly into 300 mL methanol with stirring giving rise to a stringy, fiber-like precipitate that was collected by filtration, washed thoroughly with hot water and methanol and dried under vacuum at 100 °C. Reprecipitation of the polymer from N,N-dimethylacetamide (DMAc) into methanol was carried out for further purification. The inherent viscosity of the obtained polyamide 4c was 0.87 dL g−1, measured at a concentration of 0.5 g dL−1 in DMAc at 25 °C. IR (KBr): 3180 cm−1 (amide N–H stretch), 1658 cm−1 (amide C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch). 1H NMR (300 MHz, DMSO-d6): 10.25 (s, 2H, amide N–H), 8.06 (d, J = 8.7 Hz, 4H, Hj), 7.77–7.69 (m, 6H, Ha + Hf + Hd), 7.49 (d, J = 7.3 Hz, 1H, Hi), 7.38–7.14 (m, 7H, Hg + Hh + Hk + He), 7.14–7.00 (m, 4H, Hb), 6.94 (d, J = 9.0 Hz, 1H, Hc)k, 1.37 (s, 6H, –CH3). The other polyamides were synthesized in an analogous procedure.
O stretch). 1H NMR (300 MHz, DMSO-d6): 10.25 (s, 2H, amide N–H), 8.06 (d, J = 8.7 Hz, 4H, Hj), 7.77–7.69 (m, 6H, Ha + Hf + Hd), 7.49 (d, J = 7.3 Hz, 1H, Hi), 7.38–7.14 (m, 7H, Hg + Hh + Hk + He), 7.14–7.00 (m, 4H, Hb), 6.94 (d, J = 9.0 Hz, 1H, Hc)k, 1.37 (s, 6H, –CH3). The other polyamides were synthesized in an analogous procedure.
Preparation of the polyamide films
A solution of polymer was obtained by dissolving about 0.3 g of the polyamide sample in 9 mL of hot DMAc. The homogeneous solution was poured into a 6 cm glass Petri dish, which was placed in a 90 °C oven overnight for the slow release of the solvent and then the film was further dried in vacuo at 170 °C for 12 h. The obtained films were about 35–45 μm in thickness and were used for solubility tests, X-ray diffraction measurement and thermal analyses.
Characterization
Inherent viscosities (ηinh) were measured with an Ubbelohde viscometer with a 0.5 g dL−1 of DMAc solution at 25 °C. Nuclear magnetic resonance (NMR) spectra were determined on a BRUKER-300 spectrometer at 300 MHz for 1H NMR and 75 MHz for 13C NMR in deuterated dimethyl sulfoxide. FTIR spectra were recorded by a Bruker Vector 22 spectrometer at a resolution of 4 cm−1 in the range of 400–4000 cm−1. Wide-angle X-ray diffraction (WAXD) measurements were carried out at room temperature using a Rigaku/max-rA diffractometer equipped with a Cu Kα radiation source. Differential scanning calorimetric (DSC) analysis was performed on a TA instrument DSC Q100 at a scanning rate of 10 °C min−1 in a nitrogen flow of 50 mL min−1. Thermo gravimetric analysis (TGA) was conducted with the TA 2050, with a heating rate of 10 °C min−1 under nitrogen and atmosphere. Photoluminescence (PL) spectra and fluorescent quantum yield were measured with an Edinburgh Instrument FLS920 fluorescence spectrophotometer. Fluorescent quantum yield (ΦF) was determined using a calibrated integrating sphere in NMP solution. Electrochemistry was performed with a CH Instruments 660e electrochemical analyzer. Cyclic voltammetry was conducted with the use of a three-electrode cell in which ITO (polymer films area about 0.5 × 2 cm2 prepared by spin-coating of polymer solutions (50 mg mL−1 in DMAc) onto ITO glass substrate at 1500 rpm for 60 s and then dried under vacuum) was used as a working electrode. A platinum wire was used as an auxiliary electrode. All cell potentials were taken with the use of a homemade Ag/AgCl, KCl (sat.) reference electrode. Ultraviolet-visible (UV-vis) spectra of the polymer films were measured using a Perkin-Elmer Lambda 950 spectrophotometer.
Results and discussion
Monomer synthesis
The diamine bearing fluorene-based triphenylamine units, N,N-di(4-aminophenyl)-2-amino-9,9-dimethylfluorene, was successfully synthesized by the amination reaction of 2-amino-9,9-dimethylfluorene with 4-fluoronitrobenzene followed by hydrazine Pd/C-catalytic reduction. The synthesizing routines are outlined in Scheme 1. IR and NMR spectroscopic techniques were carried out to identify the structures of the intermediate dinitro compound 1 and the target diamine monomer 2. The transformation of nitro to amino group could be monitored by the change of IR spectra (showed in Fig. S1†). The nitro groups of compounds 1 showed characteristic bands at around 1308 cm−1 and 1577 cm−1 due to NO2 asymmetric and symmetric stretching. After reduction, the characteristic bands of the nitro group disappeared and the amino group showed the typical N–H stretching absorption bands at around 3445 cm−1 and 3362 cm−1. Fig. 1 illustrates the 1H NMR spectra and 13C NMR of the diamine monomer 2, and each proton and carbon agrees well with the proposed molecular structure. The 1H NMR spectrum confirm that the nitro groups have been completely transformed into amino groups by the high-field shift of the aromatic protons and the resonance signals at around 5.0 ppm corresponding to the amino protons. Due to the introduction of the fluorene unit, the NMR spectra of the diamine 2 are very complicated. Nevertheless, the assignments of each proton can be done with the aid of two-dimensional (2D) COSY NMR spectra, as shown in Fig. S4.† Thus, the results of all the analyses suggest that the target diamine monomer 2 was prepared successfully.
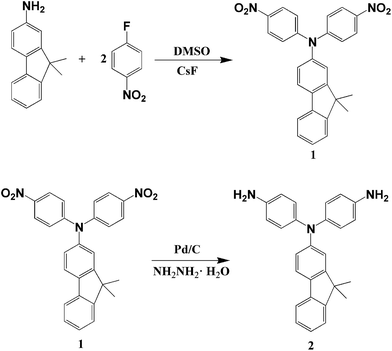 |
| | Scheme 1 Synthetic route to the diamine monomer 2. | |
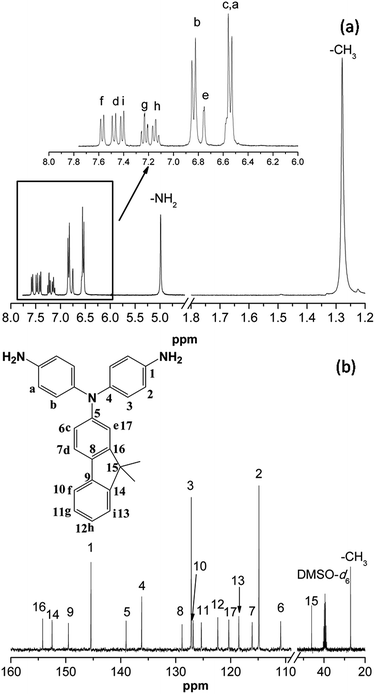 |
| | Fig. 1 (a) 1H NMR and (b) 13C NMR spectra of diamine 2 in DMSO-d6. | |
Polymer synthesis
According to the phosphorylation technique first described by Yamazaki and coworkers,23 a series of novel polyamides 4a–4e bearing fluorene-based triphenylamine units were synthesized by solution polycondensation from the diamine monomer 2 with five dicarboxylic acids 3a–3e using triphenyl phosphite and pyridine as condensing agents as shown in Scheme 2. All these polymerization reactions proceeded smoothly and afforded highly viscous polymer solution. When the resulting polymer solutions were slowly poured into methanol, all the polyamides precipitated in a tough, fiber-like form. The obtained polyamides had inherent viscosities in the range of 0.81–1.1 dL g−1 as shown in Table 2. All the polymers could afford transparent and tough films via solution casting, indicating high molecular weights. The GPC measurement of DMF-soluble polyamide 4d showed weight-average molecular weight (Mw) of 43![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 and polydispersity index (Mw/Mn) of 1.76. The formation of the polyamides could be confirmed by IR and NMR spectroscopy. The typical IR spectrum of 4a is shown in the Fig. S2,† which shows the characteristic absorption bands of the amide group at around 1658 cm−1 and 3296 cm−1. Fig. 2 shows the 1H NMR spectra of polyamide 4c and all the peaks could be assigned to the hydrogen atoms in the repeating unit. The resonance peak at 10.3 ppm clearly supports the formation of amide linkage. The IR and NMR of total polyamides can be found in the Fig. S3 and S5.† In addition, two structure related polyamides 4′c and 4′e derived from 4,4′-diamino-triphenylamine were synthesized by the same synthetic method for comparison studies.
300 and polydispersity index (Mw/Mn) of 1.76. The formation of the polyamides could be confirmed by IR and NMR spectroscopy. The typical IR spectrum of 4a is shown in the Fig. S2,† which shows the characteristic absorption bands of the amide group at around 1658 cm−1 and 3296 cm−1. Fig. 2 shows the 1H NMR spectra of polyamide 4c and all the peaks could be assigned to the hydrogen atoms in the repeating unit. The resonance peak at 10.3 ppm clearly supports the formation of amide linkage. The IR and NMR of total polyamides can be found in the Fig. S3 and S5.† In addition, two structure related polyamides 4′c and 4′e derived from 4,4′-diamino-triphenylamine were synthesized by the same synthetic method for comparison studies.
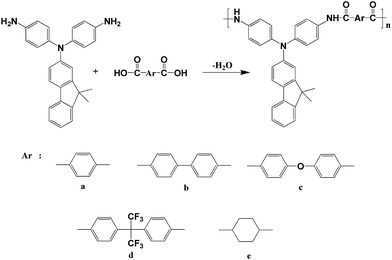 |
| | Scheme 2 Synthesis of polyamides 4a–4e. | |
 |
| | Fig. 2 1H NMR spectrum of the polyamide 4c in DMSO-d6. | |
Basic characterization
The solubility properties of polyamides were qualitatively determined at 10% w/v concentration and the results are summarized in Table 1. All the polyamides were highly soluble in polar solvents such as NMP, DMAc, DMF and DMSO. Polyamide 4d showed good solubility in less polar solvents like tetrahydrofuran (THF) and CHCl3 because of the additional contribution of the bulky hexafluoroisopropylidene fragment in the polymer backbone. Compared to the analogous polyamides 4′, these polyamides show an enhanced solubility, which can be attributed in part to the increased free-volume caused by the introduction of bulky, non-coplanar, packing-disruptive 9,9-dimethylfluorene unit in the polymer structure. Thus, the excellent solubility renders these polymers potential candidates for practical application by spin-coating or inkjet-printing processes to get large-area thin film devices.
Table 1 Solubilitya of polyamides
| Solvent |
NMP |
DMAc |
DMF |
DMSO |
THF |
CHCl3 |
| Qualitative solubility was tested with 10 mg of a sample in 1 mL of solvent. ++, soluble at room temperature; +, soluble on heating; +−, partially soluble; −, insoluble even on heating. |
| 4a |
++ |
++ |
++ |
+ |
+− |
— |
| 4b |
++ |
++ |
++ |
+ |
+− |
— |
| 4c |
++ |
++ |
++ |
+ |
+− |
— |
| 4d |
++ |
++ |
++ |
++ |
++ |
+− |
| 4e |
++ |
++ |
++ |
++ |
+− |
— |
| 4′c |
++ |
++ |
++ |
+ |
— |
— |
| 4′e |
++ |
++ |
++ |
+ |
— |
— |
The wide-angel X-ray diffraction (WXRD) patterns of these polyamides are given in Fig. S6
† indicating that the polymers are essentially amorphous, which is of great importance for their applications in flexible optoelectronic devices. Thermal properties of the polyamides were investigated by TGA and DSC and the results are summarized in
Table 2. Typical DSC and TGA (in both air and nitrogen atmospheres) curves of representative
4c are shown in
Fig. 3. All the aromatic polyamides exhibit good thermal stability with 10% weight loss temperature in nitrogen and air over 500 °C and 480 °C respectively. The carbonized residue of these aromatic polymers was up to 70% at 800 °C in nitrogen atmosphere, which could be ascribed to their high aromatic content. The glass-transition temperature (
Tg) of all these polymers were observed in the range of 256–316 °C by DSC. The lowest
Tg value of
4e is expected and can be explained in terms of the flexible cyclohexane in its backbone. All these polymers indicated no clear melting endotherms up to the decomposition temperatures on the DSC curves, which supported the amorphous nature of them characterized by WXRD. Thus, the thermal analysis results revealed that these polyamides, especially for the aromatic ones, exhibited excellent thermal stability, which will be beneficial to enhance the morphological stability to the spin-coated film and increase the service period in device application.
Table 2 Thermal properties of polyamides
| Polymer |
ηinha (dL g−1) |
Tgb (°C) |
Td10%c (°C) |
Char yieldd (wt%) |
| N2 |
Air |
| Measured at a polymer concentration of 0.5 g dL−1 in DMAc at 25 °C. Obtained at the baseline shift in the second heating DSC traces, with a heating rate of 10 °C min−1 under N2. Decomposition temperature at which a 10% weight loss was recorded via TGA at a heating rate of 10 °C min−1. Residual weight percentage at 800 °C in nitrogen. |
| 4a |
0.90 |
298 |
512 |
497 |
75 |
| 4b |
1.10 |
316 |
516 |
499 |
78 |
| 4c |
0.87 |
278 |
504 |
480 |
73 |
| 4d |
0.84 |
293 |
512 |
497 |
70 |
| 4e |
0.81 |
256 |
429 |
420 |
41 |
| 4′c |
0.76 |
271 |
531 |
500 |
71 |
| 4′e |
0.71 |
248 |
427 |
434 |
51 |
 |
| | Fig. 3 TGA and DSC (inset) curves of polyamide 4c. | |
Optical and electrochemical properties
The optical and electrochemical properties of the polyamides were investigated by UV-vis, photoluminescence spectroscopy and cyclic voltammetry. All the results can be found in Fig. S7, S8 and S10† and are summarized in Table 3. Fig. 4 depicts representative examples of the absorption and emission profiles of the polyamide 4e and 4′e in NMP at a concentration of about 1 × 10−5 mol L−1. The UV-vis absorption spectra of these polymers exhibit strong absorption bands at 334–367 nm in NMP solution, which are attributed to a π–π* conjugation transition between the aromatic rings and nitrogen atoms. In the solid film, the polyamides showed absorption characteristics similar to those in the solution with absorption onsets at 395–425 nm corresponding to optical band gaps of 2.92–3.14 eV (Eg = 1240/λonset). In the solution photoluminescence spectra, these polyamides exhibited fluorescence emission at 441–581 nm with the PL quantum yields varying from 0.37% to 47.1% as shown in Table 3. To interpret the reason why the quantum yield of the polyamide 4e was much higher than the other polyamides derived from aromatic dicarboxylic acids, the molecular simulation of two model compounds was carried out as shown in Fig. S9.† For polyamides 4a–4d, the HOMO energy level was located at fluorene-based triphenylamine moiety, while the LUMO energy level was located at phenyl-amide moiety, the low fluorescence quantum yield could be attributed to the quenching effect arising from interchain charge transfer (CT) complexing between the diphenylfluorenyl-amine donor and the phenyl-amide acceptor, which leads to a non-radiative transfer of energy. For the polyamide 4e, the excited transition from HOMO to LUMO delocalized over the fluorene-based triphenylamine, implying the locally excited (LE) transition and thus exhibited the higher quantum yield. Compared to 4′e, the higher quantum efficiency of polyamide 4e could be attributable to the presence of rigid, highly fluorescent fluorene chromophore, which has lower energy gap.
Table 3 Optical and electrochemical properties of polyamides
| Polymer |
Solutiona (nm) |
Film (nm) |
Oxidation potential (V) |
Ege (eV) |
HOMOf (eV) |
LUMOg (eV) |
| Absmax |
PLmaxb |
ΦFc (%) |
Absonset |
Absmax |
Eonset |
E1/2d |
| The polymer concentration was 10−5 mol L−1 in NMP. They were excited at the Absmax for the solution states. The quantum yield in dilute solution was determined using a calibrated integrating sphere. E1/2 (average potential of the redox couple peaks). The data were calculated from polymer films by the equation: Eg = 1240/λonset. The HOMO energy levels were calculated from cyclic voltammetry and were referenced to ferrocene (4.8 eV). LUMO = HOMO − Eg. |
| 4a |
367 |
581 |
0.41 |
424 |
366 |
0.61 |
0.76 |
2.92 |
5.10 |
2.18 |
| 4b |
367 |
560 |
0.37 |
425 |
365 |
0.60 |
0.74 |
2.92 |
5.08 |
2.16 |
| 4c |
358 |
501 |
0.68 |
399 |
359 |
0.61 |
0.74 |
3.11 |
5.08 |
1.97 |
| 4d |
361 |
525 |
0.59 |
403 |
364 |
0.64 |
0.76 |
3.08 |
5.10 |
2.02 |
| 4e |
334 |
441 |
47.1 |
395 |
331 |
0.60 |
0.73 |
3.14 |
5.11 |
1.97 |
| 4′c |
335 |
496 |
0.39 |
390 |
333 |
0.67 |
0.82 |
3.18 |
5.20 |
2.02 |
| 4′e |
316 |
384 |
5.7 |
357 |
319 |
0.63 |
0.78 |
3.47 |
5.15 |
1.68 |
 |
| | Fig. 4 UV-vis absorption and PL spectra of polyamides 4e and 4′e in NMP solutions (1 × 10−5 M). Photographs were taken under illumination of a 365 nm UV light. | |
The electrochemical properties of the polyamides were investigated by cyclic voltammetry (CV) conducted by film spin-casting on an ITO-coated glass substrate as the working electrode in anhydrous acetonitrile (CH3CN), using 0.1 M of TBAP as an electrolyte under nitrogen atmosphere. The typical CV curves for 4c and 4′c are shown in Fig. 5 for comparison. There is one reversible oxidation redox couples at half-wave potential (E1/2) value of 0.82 V (Eonset = 0.67 V) for 4′c in the first oxidation CV scan, however, after 200 cyclic scans between 0 V to 1.2 V, the polyamide 4′c gradually lost redox reversibility because of the formation of tetraphenylbenzidine (TPB) via tail-to-tail coupling. On the other hand, the polyamide 4c showed reversible oxidation redox couples at the E1/2 value of 0.74 V (Eonset = 0.61 V), and after scanning for 200 cycles between 0 V to 1.2 V, the polymer films still exhibited better electrochemical stability, meaning that the para-position of the TPA unit is protected effectively and the structure of fluorene-based triphenylamine could prevent the obtained polyamides from occurring irreversible couping reaction. The other polyamides exhibited similar CV curves to that of 4c as shown in Fig. S10† and the redox potentials of the polyamides as well as the energy of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) levels are summarized in Table 3.
 |
| | Fig. 5 Cyclic voltammetric diagrams of polyamide (a) 4′c, (b) 4c films on a ITO-coated glass substrate over cyclic scans and (c) ferrocene in CH3CN containing 0.1 M TBAP at scan rate = 50 mV s−1. | |
The HOMO levels of these polyamides were estimated from the half-wave potentials in CV experiment as 5.08–5.11 eV by using the half-wave potential of ferrocene/ferrocenium, which is 4.8 eV under vacuum. The LUMO levels were in the range of 1.97–2.18 eV estimated from the HOMO levels and the bandgaps (Eg). The closeness of the HOMO of these polymers to work function of ITO (Ef = 4.70 eV) indicates that hole injection into the molecular layer of these polymers will be effective when they are employed as hole-transporters in OLEDs.
Electro-switchable fluorescence properties
Spectroelectrochemical measurements were carried out to evaluate the optical properties in a modified fluorescence cell according to the previous report (1 cm length quartz cell) at room temperature.24 The polyamide 4e was cast on an ITO-coated glass slide used as working electrode. The electrode was placed with an angle near 45° in a 0.1 M TBAP/CH3CN solution. The sample was excited at 334 nm and the emission spectra were collected between 360 and 600 nm. As shown in Fig. 6, the fluorescence of polyamide 4e was extinguished from blue to dark, upon increasing the applied voltage from 0 V to 0.8 V. The monocation cation radical of TPA is known for acting as an effective fluorescence quencher, thus, the fluorescence is efficiently quenched. The sample returned back to its original fluorescence when the potential was subsequently set back to 0 V and the contrast ratio (If/If0) was up to 12.7. Furthermore, the stability and reversibility of the switching operation were also evaluated by repeated circulatory applied potential (0–0.8 V). As can be seen from Fig. 7, the fluorescence switching operation was reversible, reproducible and no obvious intensity changes could be observed in the first ten cycles, which can be attributed to the stable redox properties of the fluorene-based triphenylamine.
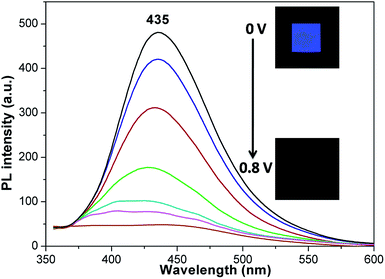 |
| | Fig. 6 Fluorescence switching of polyamide 4e at different applied potentials from 0 V to 0.8 V. The inset shows the images of the fluorescence changes under UV excitation (365 nm). | |
 |
| | Fig. 7 Fluorescence switching of polyamide 4e between 0 V and 0.8 V with a cycle time 60 s monitored at 435 nm. | |
Electrochromic properties
A homemade electrochemical cell was assembled similarly as mentioned above. The cell was placed in the optical path of the sample light beam in a UV-vis spectrophotometer to acquire electronic absorption spectra upon various applied potential. The result absorbance curves of the 4c and 4′c correlated to applied potentials are depicted in Fig. 8. In the neutral form, the colorless film of the polyamide 4c revealed obvious absorption at wavelength around 345 nm, characteristic for triphenylamine. When the applied potential increased from 0 V to 0.8 V, the intensity of absorption peak at 345 nm decreased gradually, while two new bands appeared at 425 nm and 860 nm respectively. At the same time, the color of the film changed from colorless to green. We attribute this spectral change to the formation of a stable radical cation of the fluorene-based triarylamine. When the voltage was applied in reverse, the color returned to transparent. The film colorations were distributed homogeneously across the polymer film and stable even after hundreds of redox cycles. For the comparative study, the spectral changes of the analogous polyamide 4′c (Fig. 8a) are slightly different from those of 4c (Fig. 8b), the absorption changes appeared at 405 nm, 615 nm and 834 nm with the color changed from colorless to blue.
 |
| | Fig. 8 Spectral changes of (a) polyamide 4′c and (b) 4c thin film on an ITO-coated glass substrate (in CH3CN with 0.1 M TBAP as supporting electrolyte) along with increasing of the applied voltage (vs. Ag/AgCl couples as reference). The inset shows the color changes of the polymer films between neutral and oxidized states. | |
For optical switching studies, polymer films were cast on ITO-coated glass slides in the same manner as described above, and each film was potential stepped between the bleaching (0 V) and coloring (0.8 V) state. As shown in Fig. 9, the absorbance changes at 860 nm reflect the switch in current, and the kinetics of the charge transport process can be referenced to the coloration response time. The thin film of 4c required 6.4 s at 0.8 V for coloring and 0.9 s for bleaching, which was calculated as the time reach 90% of the full switch absorbance, as it is difficult to perceive any further color change with naked eye beyond this point (Fig. S11†). After over 500 continuous cyclic scans, the polymer film still exhibited good electrochemical and electrochromic stability. The amount of Q was calculated by integration of the current density and time obtained from the graph of Fig. 9a. At the first oxidation stage, the amount of Q is 2.47 mC cm−2 for oxidation process and 2.5 mC cm−2 for reduction process. The ratio of the charge density is 98.8%, indicating that charge injection/extraction is highly reversible during the electrochemical reactions. Coloration efficiency (CE) was measured by monitoring the amount of ejected charge (Q) as a function of the change in optical density (ΔA) of the polymer films. The CEs of 4c after 500 cycles are summarized in Table 4. The CEs range from 283 cm2 C−1 for the first cycle to 226 cm2 C−1 for 500th cycle, indicating that after switching 500 times between 0 V and 0.8 V the film still retains about 80% of its optical response, therefore, these polyamides exhibit excellent electrochromic properties.
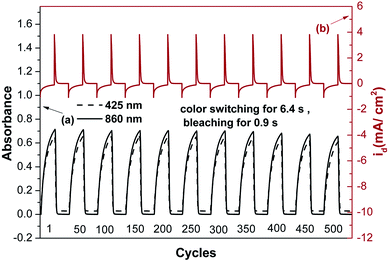 |
| | Fig. 9 (a) Potential step absorptiometry and (b) current consumption of the polyamide 4c film on the ITO-coated glass substrate (active area ∼ 1 cm2) during the continuous cyclic test by switching potentials between 0 V and 0.8 V in 0.1 M TBAP/CH3CN with a cycle time of 20 s. | |
Table 4 Coloration efficiency of polyamide 4c
| Cyclea |
ΔA860b |
Qc/mC cm−2 |
ηd/cm2 C−1 |
Decaye (%) |
| Number of cyclic switching by applying potential steps between 0 and 0.8 V (vs. Ag/AgCl), with a pulse width of 20 s. Optical density change at 860 nm. Ejected charge, determined from the in situ experiment. Coloration efficiency is derived from the equation: η = ΔA860/Q. Decay of coloration efficiency after cyclic scans. |
| 1 |
0.707 |
2.50 |
283 |
0 |
| 50 |
0.705 |
2.54 |
277 |
2.1 |
| 100 |
0.704 |
2.62 |
269 |
4.9 |
| 150 |
0.705 |
2.70 |
262 |
7.4 |
| 200 |
0.703 |
2.74 |
257 |
9.2 |
| 250 |
0.702 |
2.77 |
253 |
10.6 |
| 300 |
0.702 |
2.81 |
249 |
12.0 |
| 350 |
0.696 |
2.87 |
242 |
14.5 |
| 400 |
0.683 |
2.90 |
235 |
17.0 |
| 450 |
0.672 |
2.91 |
231 |
18.4 |
| 500 |
0.657 |
2.90 |
226 |
20.1 |
Conclusion
A new aromatic diamine monomer, N,N-di(4-aminophenyl)-2-amino-9,9-dimethylfluorene, was synthesized in high purity and high yields. A series of novel polyamides were readily prepared from the newly synthesized diamine monomer with various dicarboxylic acids via phosphorylation polyamidation reaction. Introduction of the triarylamine-based on fluorene structure to polymer main chain can not only enhance the solubility but also prevent the coupling reactions. In addition to high Tg and high thermal stability, the polymer also revealed interesting electrochromic characteristics with color changing from colorless in neutral state to green in oxidized state. Moreover, the polyamide derived from alicyclic dicarboxylic acids exhibited notable fluorescence properties and by applying reduction and oxidation potentials, the fluorescence is effectively switchable between “on” and “off” states. Thus, these polyamides may find optoelectronic applications as new electrofluorescence and electrochromic materials in the future. Further investigation of materials with this structure is currently underway by the very encouraging initial results presented here.
Acknowledgements
Financial support by the National Nature Science Foundation of China (National 973 program no. G2010CB631100) and the National Technology Support Program (no. 2011BAA06B03) are gratefully acknowledged.
References
- S. K. Deb, Appl. Opt., Suppl., 1969, 3, 192 Search PubMed.
-
(a) P. M. S. Monk, R. J. Mortimer and D. R. Rosseinsky, Electrochromism and Electrochromic Devices, Cambridge University Press, Cambridge, UK, 2007 Search PubMed;
(b) D. R. Rosseinsky and R. J. Mortimer, Adv. Mater., 2001, 13, 783 CrossRef CAS;
(c) P. R. Somani and S. Radhakrishnan, Mater. Chem. Phys., 2002, 77, 117–133 CrossRef;
(d) T. Zhang, S. Liu, D. G. Kurth and C. F. J. Faul, Adv. Funct. Mater., 2009, 19, 642 CrossRef CAS PubMed;
(e) A. Maier, A. R. Rabindranath and B. Tieke, Adv. Mater., 2009, 21, 959 CrossRef CAS PubMed;
(f) A. Maier, H. Fakhrnabavi, A. R. Rabindranath and B. Tieke, J. Mater. Chem., 2011, 21, 5795 RSC;
(g) L. Beverina, G. A. Pagani and M. Sassi, Chem. Commun., 2014, 50, 5413 RSC.
-
(a) R. J. Mortimer, Chem. Soc. Rev., 1997, 26, 147 RSC;
(b) R. J. Mortimer, Electrochim. Acta, 1999, 44, 2971 CrossRef CAS;
(c) D. R. Rosseinsky and R. J. Mortimer, Adv. Mater., 2001, 13, 783 CrossRef CAS;
(d) P. S. Somani and S. Radhakrishnan, Mater. Chem. Phys., 2002, 77, 117 CrossRef.
-
(a) P. M. Beaujuge and J. R. Reynolds, Chem. Rev., 2010, 110, 268 CrossRef CAS PubMed;
(b) P. M. Beaujuge, C. M. Amb and J. R. Reynolds, Acc. Chem. Res., 2010, 43, 1395 CrossRef PubMed;
(c) C. M. Amb, A. L. Dyer and J. R. Reynolds, Chem. Mater., 2011, 23, 397 CrossRef CAS;
(d) A. L. Dyer, E. J. Thompson and J. R. Reynolds, ACS Appl. Mater. Interfaces, 2011, 3, 1787 CrossRef CAS PubMed;
(e) E. Sefer, F. Baycan Koyuncu, E. Oguahan and S. Koyuncu, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4419 CrossRef CAS PubMed;
(f) F. Baycan Koyuncu, E. Sefer, S. Koyuncu and E. Ozdemir, Macromolecules, 2011, 44, 8407 CrossRef.
-
(a) G. S. Liou, S. H. Hsiao and H. W. Chen, J. Mater. Chem., 2006, 16, 1831 RSC;
(b) C. W. Chang, G. S. Liou and S. H. Hsiao, J. Mater. Chem., 2007, 17, 1007 RSC;
(c) Y. C. Kung, W. F. Lee, S. H. Hsiou and G. S. Liou, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2210 CrossRef CAS PubMed;
(d) H. J. Yen, K. Y. Lin and G. S. Liou, J. Mater. Chem., 2011, 21, 6230 RSC;
(e) Y. W. Chuang, H. J. Yen, J. H. Wu and G. S. Liou, ACS Appl. Mater. Interfaces, 2014, 6, 3594 CrossRef CAS PubMed;
(f) Y. Q. Wang, Y. Liang, J. Y. Zhu, X. D. Bai, X. K. Jiang, Q. Zhang and H. J. Niu, RSC Adv., 2015, 5, 11071 RSC.
- G. A. Niklasson and C. G. Granvist, J. Mater. Chem., 2007, 17, 127 RSC.
- N. Kobayashi, S. Miura, M. Nishimura and H. Urano, Sol. Energy Mater. Sol. Cells, 2008, 92, 136 CrossRef CAS PubMed.
- S. G. Lee, D. Y. Lee, H. S. Lim, D. H. Lee and K. Cho, Adv. Mater., 2010, 22, 5013 CrossRef CAS PubMed.
- R. J. Mortimer, A. L. Dyer and J. R. Reynolds, Displays, 2006, 27, 2 CrossRef CAS PubMed.
-
(a) A. Kumar, D. M. Welsh, M. C. Morvant, F. Piroux, K. A. Abboud and J. R. Reynolds, Chem. Mater., 1998, 10, 896 CrossRef CAS;
(b) S. A. Sapp, G. A. Sotzing and J. R. Reynolds, Chem. Mater., 1998, 10, 2101 CrossRef CAS;
(c) D. M. Welsh, A. Kumar, E. W. Meijer and J. R. Reynolds, Adv. Mater., 1999, 11, 1379 CrossRef CAS;
(d) I. Schwendeman, R. Hickman, G. Sonmez, P. Schottland, K. Zong, D. M. Welsh and J. R. Reynolds, Chem. Mater., 2002, 14, 3118 CrossRef CAS.
-
(a) M. A. Invernale, Y. Ding, D. M. D. Mamangun, M. S. Yavuz and G. A. Sotzing, Adv. Mater., 2010, 22, 1379 CrossRef CAS PubMed;
(b) P. M. Beaujuge, S. Ellinger and J. R. Reynolds, Nat. Mater., 2008, 7, 795 CrossRef CAS PubMed;
(c) A. Balan, D. Baran, G. Gunbas, A. Durmus, F. Ozyurt and L. Toppare, Chem. Commun., 2009, 6768 RSC;
(d) M. Icli, M. Pamuk, F. Algi, A. M. Onal and A. Cihaner, Chem. Mater., 2010, 22, 4034 CrossRef CAS.
-
(a) B. Liu, W. L. Yu, Y. H. Lai and W. Huang, Chem. Commun., 2000, 551 RSC;
(b) M. Yildirim and I. Kaya, Synth. Met., 2011, 161, 13 CrossRef CAS PubMed;
(c) K. L. Wang, M. K. Leung, L. G. Hsieh, C. C. Chang, K. R. Lee, C. L. Wu, J. C. Jiang, C. Y. Tseng and H. T. Wang, Org. Electron., 2011, 12, 1048 CrossRef CAS PubMed.
-
(a) C. D. Andrade, C. O. Yanez, L. Rodriguez and K. D. Belfield, J. Org. Chem., 2010, 75, 3975 CrossRef CAS PubMed;
(b) H. L. Wang, Z. Li, P. Shao, J. G. Qin and Z. L. Huang, J. Phys. Chem. B, 2010, 114, 22 CrossRef CAS PubMed;
(c) X. H. Ouyang and Y. P. Huo, Appl. Phys. A, 2011, 105, 891 CrossRef CAS.
-
(a) S. A. M. Ahmed, Tetrahedron Lett., 2010, 51, 730 CrossRef CAS PubMed;
(b) S. Marco and J. Andres, J. Am. Chem. Soc., 2010, 132, 8372 CrossRef PubMed;
(c) M. J. Miller, S. H. Wei, I. Parker and M. D. Cahalan, Science, 2002, 296, 1869 CrossRef CAS PubMed.
-
(a) B. J. Liu, W. Hu and C. H. Chen, Polymer, 2004, 45, 3241 CrossRef CAS PubMed;
(b) F. L. Zhang, W. Mammo and L. M. Andersson, Adv. Mater., 2006, 18, 2169 CrossRef CAS PubMed;
(c) F. L. Zhang, E. Perzon and X. J. Wang, Adv. Funct. Mater., 2005, 15, 745 CrossRef CAS PubMed.
-
(a) B. Liu, W. L. Yu and W. Huang, Maromolecules, 2002, 35, 4975 CrossRef CAS;
(b) J. Pei, X. L. Liu and W. Huang, Maromolecules, 2003, 36, 323 CrossRef CAS.
-
(a) Q. Fang, B. Xu, B. Jiang, H. T. Fu, W. Q. Zhu, X. Y. Jiang and Z. L. Zhang, Synth. Met., 2005, 155, 206 CrossRef CAS PubMed;
(b) K. Kreger, M. Bate, C. Neuber, H. W. Schmidt and P. Strohriegl, Adv. Funct. Mater., 2007, 17, 3456 CrossRef CAS PubMed;
(c) T. Miteva, A. Meisel, W. Knoll, H. G. Nothofer, U. Scherf, D. C. Muller and K. Meerholz, Adv. Mater., 2001, 13, 565 CrossRef;
(d) C. Ego, A. C. Grimsdale, F. Uckert, G. Yu, G. Srdanov and K. Mullen, Adv. Mater., 2002, 14, 809 CrossRef CAS;
(e) B. Liu and W. Huang, Thin Solid Films, 2002, 417, 206 CrossRef CAS.
- C. P. Kuo, Y. S. Lin and M. K. Leung, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 5068 CrossRef CAS PubMed.
-
(a) M. Thelakkat, Macromol. Mater. Eng., 2002, 287, 442 CrossRef CAS;
(b) Y. Shirota, J. Mater. Chem., 2005, 15, 75 RSC;
(c) Y. Shirota and H. Kageyama, Chem. Rev., 2007, 107, 953 CrossRef CAS PubMed;
(d) T. Kurosawa, T. Higashihara and M. Ueda, Polym. Chem., 2013, 4, 16 RSC;
(e) H. J. Yen and G. S. Liou, Polym. Chem., 2012, 3, 255 RSC.
- E. T. Seo, R. F. Nelson, J. M. Fritsch, L. S. Marcoux, D. W. Leedy and R. N. Adams, J. Am. Chem. Soc., 1966, 88, 3498 CrossRef CAS.
-
(a) L. Hagopian, G. Kohler and R. I. Walter, J. Phys. Chem., 1967, 71, 2290 CrossRef CAS;
(b) H. J. Yen, S. M. Guo and G. S. Liou, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5271 CrossRef CAS PubMed.
- Y. Oishi, H. Takado, M. Yoneyama, M. Kakimoto and Y. Imai, J. Polym. Sci., Part A: Polym. Chem., 1990, 28, 1763 CrossRef CAS PubMed.
-
(a) N. Yamazaki, F. Higashi and J. Kawabata, J. Polym. Sci., Polym. Chem. Ed., 1974, 12, 2149 CrossRef CAS PubMed;
(b) N. Yamazaki, M. Matusumoto and F. Higashi, J. Polym. Sci., Polym. Chem. Ed., 1975, 13, 1375 Search PubMed.
- F. Montilla, I. Pastor, C. R. Mateo, E. Morallon and R. Mallavia, J. Phys. Chem. B, 2006, 110, 5914 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra16730b |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 











