DOI:
10.1039/C5RA16683G
(Paper)
RSC Adv., 2015,
5, 92843-92857
Trends of amino acid adsorption onto graphene and graphene oxide surfaces: a dispersion corrected DFT study†
Received
18th August 2015
, Accepted 23rd October 2015
First published on 23rd October 2015
Abstract
First-principle calculations based on density functional theory were performed to investigate the adsorption properties of amino acids onto graphene surfaces. The GGA-PBE scheme with inclusion of van der Waals interactions based on a DFT-D2 approach was employed in these calculations. Several types of graphene monolayers such as perfect, defected and oxidized graphene were considered in this study and the extent and strength to which these amino acids were bound to the monolayers were explored. The accuracy of our method was validated by the state-of-the-art MP2 and hybrid B3LYP levels of theory. Based on the obtained results, the strongest interactions took place among the negatively polarized oxygen atom of the oxidized graphene and positively polarized parts of the amino acid molecules, i.e. hydrogen atoms of the –NH and –OH parts. On the other hand, the weakest interaction was attained among the delocalized π electrons from the aromatic parts of the amino acids and lone-pair electrons of the oxygen atom in oxidized graphene. Furthermore, the long range dispersion forces were found to play significant roles in the considered systems. Investigations about the role of solvation in calculations revealed that the zwitterionic conformer of glycine binds more strongly to the graphene surface in the presence of water molecules in comparison to its neutral form by an energy difference of about −4.82 kJ mol−1. The calculated geometrical parameters, binding energies and electronic structure analysis results all suggest the existence of non-covalent interactions among all of the considered species.
1. Introduction
Recent progress in understanding bio-inorganic interface interactions has opened the way for development in various scientific areas such as nanotechnology and biomedicine.1,2 Closely related to this development is the debut of carbon based nano-structures which accelerated the progress toward understanding the sophisticated nature of the physical and chemical processes involved in bio-inorganic interactions.3–5 Because of their astonishing and distinctive mechanical, electrical and optical properties, these novel materials have intrigued the scientists and drew a great deal of attention in a wide broad of scientific areas such as physics, chemistry and electronics.6–10 In recent years, bio-conjugated systems have attracted much attention in the field of biomedical applications and intense research has also been made to figure out how to employ the potential applications of these novel nanostructures in this area. A wide range of bio-molecules such as proteins and peptides were utilized to functionalize these nanostructures which are applicable in a broad range of bio-field such as molecular electronics, biochemical sensors, drug delivery as well as gene delivery.11–14 Graphene monolayer seems to be a propitious candidate for this end since it owns excellent properties such as aqueous processability and surface functionalizability.15,16 In this novel two-dimensional material which is emerged in 2004, the 2s orbital interacts with 2px and 2py orbitals to form three sp2 hybrid orbitals. Due to the exceptional electrical, thermal and mechanical properties, graphene and its derivatives have shown great potentials in various applications such as nanoelectronics, composites, sensors and medicine.17–19 Compared to carbon nanotubes (CNTs) which are the rolled form of carbon graphene sheet, the long and reactive edges of graphene make it a proper candidate for chemical modifications and metal doping.20,21 Numerous studies have reported the toxicity of CNTs which restricted their applications in bio-related fields.22,23 For example, CNTs were found to have the potential to cause inflammatory and fibrotic reactions when they reach the lungs and cannot rapidly be eliminated as a consequence of their biopersistency.24 On the other hand, graphene derivatives with smaller sizes and lower concentrations are much more biocompatible and exhibited relatively lower toxicity as compared with other materials such as CNTs.25–27
Amino acids (A.As) are the elementary units of proteins and can reflect the common chemical properties of complicated biomolecules.28 As proteins take a key role in biology, it is envisaged that understanding of their interactions with nanomaterials can resolve critical problems in the field of biomedicine. For example, investigating the adsorption mechanisms of biomolecules such as proteins on a synthetic surface may give a constructive insight to the reasons for foreign body reaction on the implanted biomaterials.29 As a powerful tool to smooth the way toward the efforts in answering numerous questions that may crop up in this direction, molecular simulation is gaining increasing popularity since it provides useful information about the structural aspects and electronic properties of molecular systems. Recently, several theoretical investigations have been performed utilizing different theoretical methods ranging from molecular mechanics to density functional theory (DFT) and even to atomic orbital based Hartree–Fock plus second-order Møller–Plesset (MP2) perturbation theory. Ganji investigated the adsorption nature of several types of amino acids onto the outer surface of single-walled carbon nanotubes (SWCNTs) and showed that these amino acids are weakly bound to the CNTs through non-covalent interactions.30 Gowtham et al. explored the adsorption nature of the five DNA and/or RNA nucleobases onto graphene and CNT and demonstrated that the nucleobases exhibit significantly different interaction strengths when physisorbed onto graphene/CNT.31,32 Using first principle calculations, Sanyal et al. have investigated the adsorption properties of DNA/RNA nucleobases on 2D transition-metal dichalcogenides and graphene sheet. They demonstrated that these nucleobases were physisorbed on the surface of graphene sheet due to van der Waals interactions and the order of binding energy of these nucleobases with graphene sheet was determined to be G > A > T > C < U.33
Despite these efforts to understand the interaction of biomolecules with these nanostructures, a comprehensive theoretical study regarding the immobilization of amino acids onto various types of graphene monolayers and the extent to which these amino acids can interact with the surface of graphene sheets has been less considered. In this work, we have investigated the interactions of three amino acids (glycine, histidine and phenylalanine) with perfect, defected graphene as well as graphene oxide (GO). Two objectives are followed in this research: (i) to explore the adsorption nature of these amino acids onto the nanostructures and (ii) to investigate the effect of structural defects and oxidized surface on the adsorption properties of the biomolecules/proteins building blocks. Detailed information about the computational procedure employed in this paper as well discussions of our obtained results are thoroughly explained through this paper.
2. Computational methods
First-principle calculations were performed based on the DFT method using OpenMX code (Open Source Package for Material eXplorer) which is based on the linear combination of pseudo-atomic orbital (LCPAO) basis function and norm-conserving pseudopotentials within local density approximation (LDA) or generalized-gradient approximation (GGA).34 For the basis set, we employed the pseudo-atomic orbitals, whose cutoff radius rc and the number of primitive orbital are summarized in Table 1. The core electrons are expressed using the norm-conserving Morrison, Bylander and Kleinman pseudopotentials and the generalized-gradient approximation (GGA) using the Perdew, Burke and Ernzerhof (PBE) functional was employed for the exchange and correlation terms.35,36 The kinetic energy cutoff was set to 150 Ry for the grid integration to determine the charge density in real space. The quasi-Newton rational function (RF)37 method was implemented for structural relaxation of all structures until the forces were less than 1 × 10−4 a.u. During the optimization procedure, all atoms were allowed to freely move and no constraints were applied. In the current calculations, a supercell composed of 84 carbon atoms was selected and a periodic boundary condition was applied. The slabs were separated from their periodic image with a 20 Å spacing in the direction perpendicular to the substrates to ensure that there was no interaction between neighboring images. A 6 × 6 × 1 Monkhorst–Pack k-point mesh was used for the Brillouin zone sampling for each system under consideration.38 Binding energies (Eb) of the adsorbates (oxygen atom and amino acids) with substrates (graphene) were calculated using the equation:
| Eb = E(complex) − [E(sub) + E(ads)] |
where E(complex), E(ads) and E(sub) are total energies of the complexes, adsorbates and substrates, respectively. We have also taken into account the long-range dispersion corrections for non-bonding vdW interactions through the DFT-D2 method proposed by Grimme.39 In order to evaluate the effect of the Basis Set Superposition Error (BSSE) in the binding energy calculation, the counterpoise correction (CP) method was used.40 To estimate the binding energy of the interacting entities with BSSE corrections, we used the following expression:
| Eb = E(complex) − [E(ads(ghost)/sub) + E(ads/sub(ghost))] |
Table 1 Cutoff radius rc of pseudo-atomic orbitals and number of the s, p and d orbitals (ns, np and nd) used in calculations
| |
H |
C |
N |
O |
| rc (a.u.) |
7.0 |
7.0 |
7.0 |
7.0 |
| ns |
2 |
2 |
2 |
2 |
| np |
1 |
2 |
2 |
2 |
| nd |
— |
1 |
1 |
1 |
The ‘ghost’ species relates to additional basis wave functions centered at the position of the adsorbates or the substrates, but without any atomic potential. The accuracy of our method for binding energies was evaluated through comparison with the state-of-the-art MP2/B3LYP level of theory.
3. Results and discussion
3.1. Adsorption of amino acids onto the perfect graphene
We first investigate the adsorption properties of glycine amino acid onto the intrinsic graphene monolayer. Based on the fact that glycine amino acid has four active sites, i.e. the amino nitrogen (N), the methyl (CH2), the carbonyl oxygen (CO) and the hydroxyl oxygen (OH), one can conclude that this amino acid tends to interact with the graphene sheet via the aforementioned active sites. Hence, several non-equivalent possible orientations were selected for the glycine molecule approaching to the surface of graphene sheet with selected active sites which are denoted as configurations G1 to G4, respectively. In addition, one additional configuration called (G5) was also considered with the molecular axis of glycine oriented parallel to the substrate. These orientation schemes are depicted in Fig. 1a–e for G1–G5, respectively. It is noteworthy to mention that for all of the considered systems, the separately optimized structures of substrate and amino acids were used to construct the configurations. The optimized structures and geometrical parameters of all amino acids and substrate are presented in ESI (Fig. S1†). Since we set out to obtain the approximate adsorption configuration of the system, we calculated Single Point Energy (SPE) for all of the considered configurations and tabulated the results. Following this purpose, we have fixed the structures of the amino acid and the graphene monolayer and translated the amino acid vertically toward the surface of the substrate and performed SPE calculations for all of the orientations. Then the binding energy of the system as a function of separation distance between two closest atoms of these molecules was calculated. For a better description of the involved procedure and better understanding of the obtained results, we have included the single point energy plot which can be seen in ESI (Fig. S2†). From the obtained SPE plot, we have concluded that altering the initial structure, distance and orientations of the adsorbate can affect the results so that the calculated binding energies varied as the amino acid approached the substrate with different active sites and different distances. Moreover, as the adsorbate molecule approached too close to the surface of the substrate, the interactions became rapidly repulsive as the consequence of nucleus–nucleus repulsions which confirms that the separation distance can also affect the final result. The lowest obtained binding energies at each orientation and their respective equilibrium distances are tabulated in Table 2. In addition, we have also carried out a minimum energy scan in the energetically most favorable structure i.e. G5, to find possible lower energy orientations. For this end, we have fixed the structures of the adsorbate and substrate in G5 configuration at the distance corresponding to its equilibrium value in Table 2 and rotated the glycine molecule around the X axis by 30° through 360° (12 steps). Then, we have performed single point energy calculations at each step and plotted the energy landscape for the system as a function of the rotation angle of the glycine molecule above the graphene monolayer (see Fig. 3).
 |
| | Fig. 1 (a)–(e) Orientation schemes for a glycine molecule approaching the hexagonal network of the graphene sheet with its respective active sites. (f)–(h) Optimized structures of graphene/glycine complex, coronene-like and coronene-like/glycine complex, respectively (red: O, blue: N, gray: C and white: H). | |
Table 2 Binding energies and equilibrium distances resulted in SPE calculations for graphene/A.A complexes. The value in parentheses denote the adsorption energy of the most stable configurations after full structural optimization but without BSSE corrections
| Configurations |
Eb (eV) |
Distance (Å) |
| Glycine |
| G1: NH2 |
−0.20 |
3.296 |
| G2: CO |
−0.17 |
3.133 |
| G3: OH |
−0.12 |
3.127 |
| G4: CH2 |
−0.19 |
2.684 |
| G5: parallel |
−0.22 |
3.505 |
| G5@180° |
−0.33 (−0.49) |
2.994 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
| Histidine |
| G6: CH3 |
−0.18 |
2.838 |
| G7: N |
−0.23 |
3.231 |
| G8: ring |
−0.51 (−0.63) |
3.405 |
|
| Phenylalanine |
| G9: RH + CH2 |
−0.53 |
2.944 |
| G10: RH2 + CH2 |
−0.55 |
2.916 |
| G11: ring |
−0.61 (−0.78) |
3.361 |
We next performed full structural optimization procedure for the most favorable structure corresponding to the lowest obtained energy among all orientations, i.e. G5@180° configuration. As it is shown in Fig. 1f which is the optimized structure of the energetically most stable configuration, the glycine molecule has parallel orientation with respect to the planar surface of graphene sheet and hydrogen atoms of its amino group are directed toward the surface. The binding energy for the energetically favorable configuration is about −0.39 eV with the BSSE corrections and the equilibrium distance between the closest atoms of the glycine molecule with respect to graphene sheet is about 2.514 Å. Furthermore, the geometrical parameters of the glycine molecule underwent a very small change upon the adsorption onto the graphene. These results suggest that the glycine molecule is weakly bound to the surface of the graphene sheet having a binding energy which is typical for the physisorption.41,42
To estimate the reliability of our reported values, we have performed benchmark calculations within the framework of state-of-the-art hybrid B3LYP method, Beck's three-parameter exchange functional and Lee, Yang, Par correlation functional with the def2-TZVP (Ahlrich's split-valence triple zeta plus polarization) basis functions.43 Furthermore, the more accurate version of Grimme's atom pair-wise dispersion correction, so-called DFT-D3 method, was also incorporated in our calculations.44 All-electron benchmark calculations were carried out using the ORCA quantum-chemistry program45 to determine the electronic structures and binding energies. Since the supercell technique is not implemented in ORCA quantum-chemistry software and periodic boundary conditions cannot be applied in this program, we selected a graphene flake constructed by a slab of hexagonal lattice consisting of 54 carbon atoms terminated by hydrogen atoms, so-called coronene-like, to represent the graphene sheet and allowed to fully relax to reach an equilibrium state. Numerous studies demonstrated that the coronene-like surface serves to be a suitable model to represent the graphene sheet and can be used alternatively instead of the periodic graphene in programs which are not capable of handling periodic systems.46–48 Schematic representations of the coronene-like molecule with relaxed structure is shown in Fig. 1g. Similar to previous section, different initial configurations were selected for a glycine molecule approaching the surface of the coronene-like with its possible active sites and similarly SPE calculations were performed to find the approximate adsorption configuration. Then, the structure with the lowest obtained energy was considered as the most favorable structure and fully optimized. The obtained results show that glycine molecule prefers to be adsorbed onto the coronene-like surface having parallel orientation and with binding energy of −0.394 eV with BSSE corrections and the distance between two closest atoms of the glycine molecule and coronene-like was determined to be 2.675 Å (see Fig. 1h). We find that B3LYP results confirm our current results obtained by DFT-D2 method within the framework of GGA-PBE scheme. Hence, we take advantage of this method for further investigations about the electronic properties of the system and the binding nature between the glycine molecule and graphene sheet.
We have also made inquiries about the interaction of aromatic amino acids with the graphene sheet. For this purpose, we have employed histidine and phenylalanine molecules as the aromatic representatives of twenty most common amino acids. Considering the fact that the twenty most common amino acids first and foremost differ through their respective side chains, we decided to concentrate only on these sections and eliminate their peptide backbones by terminating hydrogen. Hence, the histidine molecule is represented by an imidazole ring attached to a methyl group and phenylalanine molecule is represented by toluene. One of the positive outcomes of this technique is that one can achieve reconciliation between accuracy of the results and computational efficiency. Throughout this study, these side chains were mentioned by their equivalent amino acid appellations. It is well-known that non-covalent interactions play a major role in describing many biological problems where these interactions are mainly governed by weak vdW dispersion forces. The key challenge is to find suitable theoretical method to accurately describe these interactions. Recently, Mollenhauer and coworkers have investigated the performance of different DFT approaches in combination with dispersion corrections and revealed that the DFT-D2 method with the GGA-PBE scheme can excellently describe the interactions of aromatic molecules with extended carbon based systems.49 Throughout this work we similarly employ DFT-D2 method within the GGA-PBE functional to investigate the interaction of aromatic amino acids with the graphene monolayers. In order to examine the interactions of these aromatic amino acids with graphene sheet, several initial configurations were selected as the possible favorable configurations and similar computational procedure was performed to approximately determine the energetically most favorable structures. These configurations are denoted from (G6) to (G8) for histidine molecule and (G9) to (G11) for phenylalanine molecule approaching the hexagonal network of the graphene (see Fig. S3 in ESI†). Results of SPE calculations are also tabulated in Table 2. After full structural optimization of configurations which own the lowest energy, we have figured out that these aromatic amino acids prefer to be adsorbed on graphene sheet with parallel orientation and their respective aromatic rings were found to interact with the hexagon of graphene in the form of AB stack. The optimized structures of histidine and phenylalanine molecules adsorbed on the hexagonal network of the graphene are provided in ESI (Fig. S4†). The calculated binding energies were found to be −0.51 and −0.61 eV with BSSE corrections and the average inter-planar distances were 3.231 and 3.364 Å for histidine and phenylalanine, respectively. These results are in good agreement with those obtained by Majumder et al. which explored the interactions of aromatic amino acids with graphene sheet and CNT at the MP2/6-31G* level of theory as implemented in GAUSSIAN 03 program.50 Furthermore, the geometrical parameters of these amino acids such as the bond lengths and angles remained almost unchanged after the binding process. Mulliken charge analysis revealed that 0.03 and 0.06e of charge were transferred from the graphene sheet to the histidine and phenylalanine molecules, respectively. The obtained binding energies and equilibrium distances all suggest the existence of rather strong physisorption between these amino acids and the graphene sheet.51–53 These interactions are attributed to π–π interactions.
It has been previously mentioned that in order to reconcile between the accuracy of the results and computational efficiency, we have eliminated the peptide backbone and just concentrated on the side chains. To evaluate the accuracy of this technique and also to check if any noteworthy deviations can occur in final result, the complete L-histidine molecule was located above the graphene monolayer and allowed to fully relax to reach an equilibrium state. After full structural optimization of the considered system, the imidazole ring was positioned above the hexagon of the graphene in the form of AB stack with the average equilibrium distance of 3.185 Å. Furthermore, energy calculation result revealed that the L-histidine molecule bound to the surface of the graphene sheet with an energy value of −0.527 eV with the BSSE corrections. Obtained results show a good consistency with those obtained by simplified histidine molecule which indicates that the peptide backbone does not make any significant alterations in the final results. The optimized structure and geometrical parameters of the system is illustrated in ESI (Fig. S5†).
It is well known that conventional density functionals are not capable of accurately predicting the interaction energies in biological systems since they do not account for long range vdW interactions. Since in these systems, interactions such as hydrogen bonding and stacking are crucial, thus the dispersion forces tend to have significant impact on the binding energies and electronic structures of these systems and outperform the conventional DFT. In order to explore the role of long range dispersion forces in the systems under study, we have constructed three orientation schemes identical to G5, G8 and G11 (the energetically most favorable configurations based on previously obtained results) and fully optimized them without the inclusion of vdW corrections. The separately optimized structures of graphene and A.As with the conventional DFT were used to construct these orientation schemes. After full structural optimization of these systems, dramatic changes in the binding energies were observed. The binding energies were calculated to be about +0.12, +0.10 and +0.14 eV with the BSSE corrections, respectively. Furthermore, the equilibrium distances between the closest atoms of the two entities, i.e. A.As and graphene, notably increased. The average equilibrium distances of these A.As with graphene sheet were measured to be 3.1, 3.54 and 3.7 Å for glycine, histidine and phenylalanine molecules, respectively. From the obtained results, we found that vdW forces have significant effect on the energetic and geometrical parameters of these systems.
We also determined the electronic density of states (DOS) of the graphene/A.A complexes to fully understand the bonding nature of these amino acids through electronic structure. It can be seen from Fig. 2 that in all the cases, the electronic spectra near the Fermi level remained unchanged after the adsorption process. Indeed, the energy spectra for the graphene/A.As are almost the same as those for perfect graphene and no significant change can be observed on the DOS near the Fermi level upon the adsorption of amino acids onto the graphene sheet. Moreover, the DOS near the valence band underwent a distinct change compared to that of perfect graphene which shows that some local energy appeared after the adsorption process and resulted in a decrease the energy gap. To gain deeper insight into the interaction nature between these amino acids and graphene sheet, we also calculated total density map of the combined systems. As it is shown in Fig. 4, for all the cases, the amino acids positioned far from the graphene monolayer and the electronic charge distributions across the C atoms remained unaltered which indicates that no significant charge transfer was occurred between two species and validates the results provided by Mulliken charge analysis and DOS curves.
 |
| | Fig. 2 Total density of states of: (a) graphene/glycine, (b) graphene/histidine and (c) graphene/phenylalanine complexes. | |
 |
| | Fig. 3 Schematic representations of: (a) glycine molecule above the graphene monolayer after 180° rotation, (b) energy landscape plot as a function of the rotation angle of the glycine molecule above the graphene monolayer. | |
 |
| | Fig. 4 Isosurface maps of the total charge density for the graphene/A.A complexes with isosurface value of 0.07 a.u. ((a) and (b) top and side views of the total density maps for glycine/graphene, (c) and (d) top and side views for graphene/histidine and (e) and (f) top and side views for graphene/phenylalanine, respectively). | |
3.2. The role of structural defects on the adsorption properties
We now consider the adsorption nature of amino acid molecules onto a defected graphene using similar computational procedure. Structural defects are naturally occurring defects that alter the structure of sp2 bonded carbon nanomaterials and play crucial role in formation, transformation and electrical properties of carbon nanostructures. In order to understand the role of structural defects in the adsorption properties of amino acids onto the surface of graphene sheet, we embedded a Stone–Wales (SW) defect on the hexagonal network of the planar graphene. The schematic representation of this defect and its respective possible adsorption sites are illustrated in Fig. 5a. Similar to previous section, several initial configurations were selected for the amino acid molecules approaching the center of pentagon, hexagon and heptagon ring of the defected graphene sheet via their respective active sites. SPE calculations were carried out to approximately find the configuration which is close to the favorable structure. Then, the structure with the least energy was allowed to fully relax to reach an equilibrium state. Several possible configurations were selected with the imidazole ring, nitrogen atom of the imidazole ring and methyl group attached to the histidine molecule, approaching directly above the center of the hexagon, defected site pentagon and heptagon of the graphene sheet (configurations D1–D9). Likewise, similar orientations were also selected for glycine molecule and phenylalanine with their expected active sites interacting with the mentioned defected sites of the graphene (configurations D10–D24 for glycine molecule and D25–D33 for phenylalanine). The lowest obtained binding energies at each orientation and the distance between two closest atoms of the defected graphene and amino acids are given in ESI (Table S1†).
 |
| | Fig. 5 Optimized structures and geometrical parameters of: (a) defected graphene, (b) defect/glycine, (c) defect/histidine and (d) defect/phenylalanine complexes (red: O, blue: N, gray: C and white: H). | |
Our calculated results show that these amino acids are physisorbed on the defected site of the graphene sheet with the binding energies of −0.37, −0.49 and −0.61 eV with BSSE corrections for glycine, histidine and phenylalanine molecules, respectively. Furthermore, after performing full structural optimization procedure for the configurations with lowest binding energy, the geometrical parameters for all of the considered amino acids remained almost unaltered during the adsorption process. It can be found from the calculated binding energies that in all the cases the defect caused a slight decrease in the interaction between the two entities in comparison to that of perfect graphene. The optimized structures of configurations with the lowest binding energy are shown in Fig. 5. Mulliken population analysis revealed that 0.07, 0.02 and 0.04e of charge were transferred from the defected graphene sheet to glycine, histidine and phenylalanine molecule, respectively. All of the above findings confirm the existence of a weak interaction between the defected graphene and amino acids and show that the adsorption capability of perfect graphene is slightly better that that of defected one.
3.3. Adsorption of amino acids onto the graphene oxide (GO)
Following our attempts to comprehensively cover a wide breadth of aspects that could alter the sensitivity of carbonic hexagonal monolayers toward biomolecules we have functionalized the graphene sheet by an oxygen atom and explored the interactions of amino acids with this modified monolayer surface as a simple prototype for the so-called graphene-oxide (GO).54 Since the discovery of graphene oxide in 2004, this structure has attracted very much attentions in several fields due to its remarkable properties such as water dispersity,55 antibacterial activity56 and amphiphilic characteristics.57 Moreover, it has many potential applications such as biochemical sensing,58 cell imaging59 and drug delivery60 which have been thoroughly investigated in literature. For this purpose, we first considered the adsorption nature of an oxygen atom onto a graphene monolayer. The oxygen-decorated graphene was modeled by a single oxygen atom per unit cell which is initially located on three high symmetric adsorption sites, i.e. top site directly above a carbon atom, bridge site above the C–C bond between the two carbon atoms and the hollow site, above the center of the hexagon (see Fig. 6a). After full structural optimization of the considered structures, we found that in all the cases, the oxygen atom moves toward the bridge site with an equilibrium distance of 1.47 Å which indicates that the bridge site is the most energetically favorable location for the oxygen atom to be adsorbed.61 Fig. 6b illustrates the optimized structure of the oxygen-decorated graphene. The calculated binding energy with BSSE corrections for the most favorable configuration is about −4.88 eV which is in good agreement with other theoretical works and seems high enough to be considered as chemisorption.62–64 To explore the nature of the adsorption and to get further insight into the electronic properties of the system, we have also performed DOS curve for the oxygen-decorated graphene and compared the results with those for perfect graphene. It can be seen from the Fig. 6c that the DOS spectra near the Fermi level have obvious changes due to the adsorption of oxygen atom. Indeed, the adsorption of oxygen atom leads to substantial perturbations near the Fermi level as a result of hybridization process between respective orbitals. Furthermore, the Fermi level of the graphene shifted about 0.12 eV toward the lower energy region after the adsorption of oxygen atom which is attributed to charge transfer. Mulliken charge analysis revealed that 0.16e of charge was transferred from the graphene to oxygen atom. All of the above mentioned results indicate the existence of strong interaction between the oxygen atom and the graphene sheet which confirms the above observation regarding the nature of the adsorption process.
 |
| | Fig. 6 Schematic representations of: (a) possible adsorption sites for the oxygen atom onto the surface of the graphene, (b) optimized structure and geometrical parameters of the oxygen-decorated graphene and (c) total density of states for the most favorable adsorption configuration (red: O, gray: C). | |
To investigate the interactions of amino acids with this functionalized monolayer, several initial configurations were selected and SPE calculations were carried out to approximately determine the most stable configurations. Since the oxygen atom of the graphene sheet is highly electronegative and also contains lone-pair of electrons, it is partially negatively charged. On the other hand, the hydrogen atoms attached to highly electronegative atoms are positively polarized. Thus, among all possible configurations only those were selected that their respective active sites were susceptible to attractive electrostatic interaction but of varying strength. Hence, three different configurations named O1 to O3 were selected for the glycine molecule approaching the oxygen of the graphene sheet with the hydrogen atoms of –OH, –NH2 and –CH2 group, respectively. Likewise, five non-equivalent configurations were also selected for each of the histidine and phenylalanine molecules approaching the oxygen atom of the graphene with their respective active sites. These configurations are denoted from O4 to O8 for the histidine and O9 to O13 for phenylalanine. Results of SPE calculations are given in Table 3. Then full structural optimization of configurations with the lowest SPE was carried out. The optimized structures are depicted in Fig. 7. As it can be seen from the figures, no significant changes can be observed in geometrical parameters of the glycine and phenylalanine molecule and their bond lengths remained almost unchanged during the adsorption process. For histidine molecule, the –NH bond slightly elongated after the adsorption process which tells us about the existence of a rather strong interaction between histidine and oxygen decorated graphene. Energy calculation results revealed that these amino acids bound to the surface of the oxygen decorated graphene within an energy range which is typical for physisorption. Calculated binding energies are −0.44, −0.71 and, −0.31 eV with BSSE corrections for glycine, histidine and phenylalanine, respectively. It can be seen from the results that the binding energy of the glycine and histidine molecule is evidently higher than those for perfect graphene. On the other hand, the binding energy of the phenylalanine underwent a significant decrease in comparison to that of perfect graphene. The increase in the binding energy for the glycine and histidine molecules can be attributed to the strong electrostatic attractions between the positively charged hydrogen atoms of these amino acids and negatively charged oxygen atom of the graphene. In the case of phenylalanine, however, the decrease in binding energy stems from the electrostatic repulsion between oxygen lone-pair electrons and delocalized π electrons of the benzene ring.
Table 3 Binding energies and equilibrium distances resulted in SPE calculations for GO/A.A complexes. The value in parentheses denote the adsorption energy of the most stable configurations after full structural optimization but without BSSE corrections
| Configurations |
SPE (eV) |
Distance (Å) |
| Glycine |
| O1: CH2 |
−0.12 |
3.296 |
| O2: NH2 |
−0.14 |
3.133 |
| O3: OH |
−0.26 (−0.54) |
3.127 |
|
| Histidine |
| O4: CH3 |
−0.06 |
2.684 |
| O5: NH |
−0.24 (−0.85) |
2.240 |
| O6: ring |
−0.18 |
2.838 |
|
| Phenylalanine |
| O7: RH + CH2 |
−0.13 |
3.231 |
| O8: RH2 + CH2 |
−0.05 |
3.405 |
| O9: ring |
−0.23 (−0.43) |
2.944 |
 |
| | Fig. 7 Optimized structures and geometrical parameters of: (a) GO/glycine and (b) GO/histidine complexes. (c) and (d) Denote to the top and side views of the GO/phenylalanine complex after geometrical relaxation (red: O, blue: N, gray: C and white: H). | |
To further investigate the adsorption nature of these amino acids onto the graphene-oxide, we have also plotted DOS curves for the combined systems and compared the results with the energy spectra obtained from individual parts of the system i.e. the functionalized graphene and amino acids (see Fig. S6 in ESI†). It can be seen from the figures that in all the cases, the energy spectra near the Fermi level remained unaltered after the adsorption process and there is no evidence of hybridization between respective orbitals of the system. Furthermore a minor blue-shift was observed in the Fermi level of the functionalized graphene when amino acids were bound it. The Fermi levels of the glycine, histidine and phenylalanine shifted about 0.03, 0.08 and 0.01 eV toward the lower energy region, respectively. These slight decreases in the Fermi energy suggest that an insignificant charge transfer took place from the graphene sheet to the amino acids. Mulliken charge analysis showed that 0.05, 0.06 and 0.01e of charge was transferred from the oxygen decorated graphene to the glycine, histidine and phenylalanine molecules, respectively. The amount of transferred electrons and the direction in which the charges are transferred emphasizes the accuracy of the results obtained by DOS curves and confirm the hypothesis regarding the nature of the adsorption. For a better demonstration of the binding nature between oxygen decorated graphene and amino acids, we have also calculated the total density map for the most stable structures and presented the results in ESI (see Fig. S7†). It can be observed from the figures that in all the cases, the adsorbed amino acids are positioned far from the graphene monolayer and have no effect on the electron distribution pattern across the C and O atoms of the sheet. Hence, there is no evidence of hybridization between two closest atoms of the graphene sheet and amino acids and no significant charge transfer occurs in the adsorption.
3.4. Adsorption properties for zwitter ionic case and the solvent effect
It is well established that the most outstanding properties of amino acids are due to their amphoteric nature because of the existence of two functional groups of different polarities on their structure: the –NH2 and the –COOH functional groups. These functional groups are, for example, responsible for the existence of a tautomeric equilibrium between the neutral form and the zwitter ionic form of the amino acids.65 In the gas phase, the neutral form of the amino acids is the most stable conformer but with a change in the dielectric of the medium, for example in a polar solvent such as water, the zwitter ionic form will be energetically favorable. In this section, we have prepared a brief report about the changes in the adsorption nature of the amino acids upon changing the dielectric constant of the medium. In principle, a complete understanding of the involving processes associated with the interactions of amino acids with carbonic monolayers in polar solvents should include all 20 most common amino acids which, obviously, is computationally impractical, especially with the level of accuracy we have employed in our computations and requires separate studies. For this reason, we have only selected the glycine molecule as the simplest of all the amino acids, which can be served as a prototype and the initial point, to evaluate the interactions of biomolecules with carbonic monolayers in an aqueous environment. Following this purpose, we have employed the Conductor like Screening Model (COSMO) as implemented in ORCA quantum chemistry program at the B3LYP-D3/def2-TZVP level of theory to explore the role of solvation effects in the adsorption properties and geometrical parameters of the system. The so-called coronene-like model was used to represent the graphene sheet in our study. Furthermore, two conformers of the glycine molecule, the neutral form and the zwitter ionic form, were also selected to study the behavior of the system within a polar solvent. It should also be noted that, throughout this section, a dielectric constant of 78.39 was incorporated in our calculations which corresponds to the experimental value for liquid water at room temperature. We first carried out structural optimization procedure for the mentioned conformers of the glycine within the gas phase and investigated the stability of the system based on the obtained total energy. After full structural optimization of the system, we noticed that the neutral conformer is energetically more stable than the zwitter ionic form. Interestingly, the zwitter ionic conformer does not seem to exist in the gas phase as a stable conformer. Indeed, after structural optimization, this structure underwent proton transfer and moved to the lower energy neutral structure. These findings are in consistency with previous studies which mentioned that at least two water molecule,66 a crystal field or a continuum solvent distribution67 is necessary to stabilize the zwitter ionic form in gas phase and to prevent the proton transfer. Schematic representations of the zwitter ionic form of the glycine molecule before and after optimization in gas phase are shown in Fig. S8(a and b).† In the next step, we have optimized the structures of these conformers in the presence of water molecules and evaluated the stability of them in terms of the obtained energy and geometrical parameters. Fig. S8(c and d)† illustrates the optimized structures of these molecules. From this figure it can be seen that the zwitter ionic form preserved its structure and proton transfer process did not happen. Moreover, an energy difference of about −26.5 kJ mol−1 was also observed in the obtained energy which confirms that the zwitter ionic form is energetically more favorable than the neutral conformer within a polarized solvent. By summing all the obtained results, we have concluded that within a polarized environment, the zwitter ionic form of the glycine is the dominating structure, while the neutral conformer is the favorable structure as the solvent effects are eliminated in calculations. In light of understanding the adsorption properties of glycine molecule onto the graphene monolayer within a polar solvent, we have investigated the both mentioned conformers of the glycine interacting with graphene monolayer and constructed several configurations with respect to their different active sites. Several studies demonstrated that the geometrical parameters of the system does not change significantly in comparison to the gas phase as the solvent is incorporated into the calculations.68,69 Moreover, the optimized structures of the neutral glycine in the presence and absence of water, which was presented earlier in this section, was almost the same and no significant differences were observed between two respective systems. As a result, to explore the effect of solvation in the adsorption properties of the neutral glycine onto the graphene monolayer, we have used the optimized geometry of our benchmark system in Section 3.1 and performed single point energy calculations for these structures (complex, coronene-like and glycine molecule) in the polarized medium to obtain the binding energy. Energy calculation result revealed that the glycine molecule interacts with the graphene surface with a binding energy of −0.399 eV with the BSSE correction in the presence of water molecules in the environment. Comparison of the obtained result in the solvent with those obtained in the gas phase shows that the binding energy of the system undergoes a slight increase upon the change in the dielectric of the medium which suggests that the presence of solvent results in some inconsiderable improvements in the interactions of the neutral glycine with the graphene sheet.
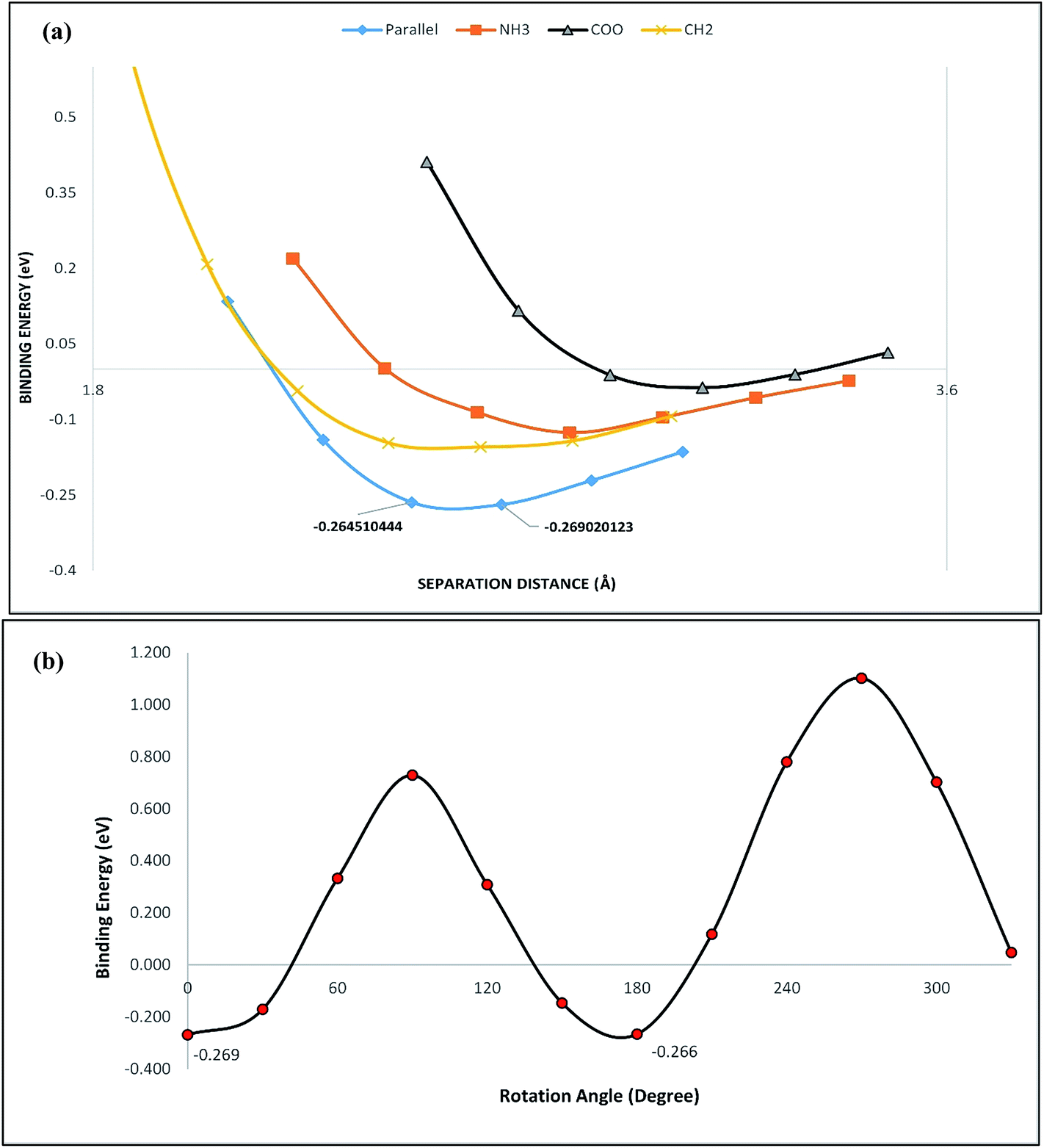
We next have investigated the interactions of the zwitter ionic form of the glycine with the graphene monolayer in the vicinity of water molecules. For this end, four non-equivalent possible configurations were selected for the zwitter ionic glycine approaching the graphene monolayer with its possible active sites, i.e. –COO−, CH2 and NH3+ functional groups. In addition, on additional configuration was also considered for the zwitter ionic glycine with parallel orientation, positioned above the graphene monolayer. These configurations are presented in Fig. 8. In order to find the approximate adsorption configuration we have fixed the structure of the zwitter ionic glycine and graphene monolayer and translated the glycine molecule vertically toward the surface in a step of 0.2 Å from its initial distance. At each step, single point energy calculations were carried out to obtain the binding energy as a function of the separation distance between the closest atoms of glycine and graphene sheet. Fig. 9a shows the SPE plot for these four orientations. We have also scanned for other possible lower energy orientations in the energetically most favorable configurations i.e. parallel configuration. Following this purpose, we have fixed the structure of parallel configuration and rotated the glycine molecule around the Y-axis in a step of 30° through 360° (12 steps) and performed single point energy calculations to obtain the binding energies. From the obtained energy landscape which is shown in Fig. 9b, it can be seen that the initial orientation of the parallel configuration was the energetically most stable and rotations of the glycine molecule resulted in binding energies that were lower than the initial one. Then, full structural optimization procedure was performed to the most favorable configuration in which the structure was allowed to fully relax to reach an equilibrium state. The optimized structure of the parallel configuration is illustrated in Fig. 10. Obtained results showed that the zwitter ionic glycine prefers to be adsorbed on the surface of the graphene sheet with the binding energy of −0.45 eV with BSSE corrections and equilibrium distance of 2.544 Å between the closest atoms of the glycine and graphene sheet. The obtained binding energy and the equilibrium distance suggest the existence of a rather strong interaction between the two species which is typical for physisorption. Comparing the results with those obtained by neutral glycine shows that the zwitter ionic conformer of the glycine interacts rather more strongly with the graphene monolayer and the formed complex between the two molecules is thermodynamically more stable in the presence of a polar solvent in comparison to the formed complex in gas phase. Moreover, amino acids are found to exist dominantly in zwitter ionic form in neutral water and the neutral form of amino acids in such media is so negligible that their mole fractions can hardly be measured through experimental measurements. As a consequence, it was expected for the zwitter ionic glycine to bound more strongly to the graphene surface in comparison to the neutral form within the water solution which confirms our conclusions regarding the stability of the formed complex.
 |
| | Fig. 8 Schematic representations of the zwitter ionic glycine approaching the hexagon of the graphene with different active sites: (a) CH2, (b) COO−, (c) NH3+ and (d) parallel. (e) Represents the X and Y-axis of the system (red: O, blue: N, gray: C and white: H). | |
 |
| | Fig. 9 (a) SPE plot for the zwitter ionic/graphene complexes. (b) Energy plot as the function of rotation angle of the zwitter ionic glycine around the Y-axis. | |
 |
| | Fig. 10 Optimized structure and geometrical parameters of the zwitter ionic glycine/graphene complex within the solvent (red: O, blue: N, gray: C and white: H). | |
4. Conclusions
In light of understanding the interactions of graphene derivatives with larger biomolecules and the extent to which these monolayers can be biofunctionalized, we have performed first-principle calculations based on density functional theory (DFT) to investigate the adsorption nature of glycine, histidine and phenylalanine onto the graphene monolayers. Although larger biomolecules are much more complicated than these amino acids, however all of them include amino nitrogen, carboxyl oxygen, hydroxyl oxygen, imidazole and aromatic rings as their possible active sites. Hence, the binding properties and the interaction natures of larger biomolecules such as proteins with graphene derivatives can be reasonably extrapolated from the results obtained in this paper. Following our objectives in this study, we have explored the adsorption nature of several amino acids on perfect, defected and oxidized graphene monolayer. Results demonstrated that for all the cases, the binding energy values lied in the range of physisorption but of varying strength. Furthermore, the upshots of the DOS curves and total density map confirmed the existence of non-covalent interactions as a consequence of weak but numerous van der Waals forces among considered species. The strongest interactions took place among the positively polarized part of the amino acids and negatively charged oxygen of the graphene oxide as a consequence of electrostatic attractions. On the other hand, the weakest interactions occurred between delocalized π electrons of aromatic rings and the lone-pair electrons of oxygen atoms. In the case of the perfect graphene, the strongest interactions occurred between the aromatic parts of the amino acids and the hexagonal network of the graphene which can be attributed to π-stacking. Obtained results regarding the effect of a change in the dielectric of the medium suggest that the zwitter ionic conformers of amino acids are more stable in the presence of a polar solvent such as water and form rather more stable complexes with carbonic monolayers in such media in comparison to their neutral form. However, to draw a general conclusion regarding the behavior of amino acids within a polar solvent and their interactions with carbonic monolayers, more detailed studies are mandatory and, obviously, this subject has room for further research. Therefore, from the obtained results one may get detailed information to decipher the processes which govern the interactions between bio-inorganic interfaces and conclude that similar to perfect graphene, oxygen decorated graphene may also act as a potential candidate for biofunctionalization and other bio-related applications. Meanwhile, further studies are also needed to comprehensively investigate the interactions of biomolecules with other types of 2D monolayers such as gallium-nitride graphene (GaN), aluminium-nitride graphene (AlN) and other types of group III-nitride monolayers as proper candidates which can be extensively used in bio-related applications. In addition, the role of solvation effects in the interactions of these biomolecules with different 2D monolayers should also be addressed thoroughly in future works.
Acknowledgements
The authors gratefully acknowledge the support of this work by Babol Noshirvani University of Technology and Pharmaceutical Sciences Branch, Islamic Azad University (IAUPS). We also acknowledge the Foundation of Supercomputing Center of Nanotechnology Research Institute (NRI), Babol Noshirvani University of Technology-Iran.
References
- C. Sanchez, K. J. Shea and S. Kitagawa, Chem. Soc. Rev., 2011, 40, 471–472 RSC.
- S. Mukhopadhyay, R. H. Scheicher, R. Pandey and S. P. Karna, J. Phys. Chem., 2011, 2, 2442–2447 CAS.
- S. Iijima, Nature, 1991, 354, 56–58 CrossRef CAS.
- S. Iijima and T. Ichihashi, Nature, 1993, 363, 603–605 CrossRef CAS.
- O. Shenderova, V. Zhirnov and D. Brenner, Crit. Rev. Solid State Mater. Sci., 2002, 27, 227–356 CrossRef CAS.
- D. M. Guldi and V. Sgobba, Chem. Commun., 2011, 47, 606–610 RSC.
- X. An and C. Y. Jimmy, RSC Adv., 2011, 1, 1426–1434 RSC.
- S. Zhu, J. Zhang, C. Qiao, S. Tang, Y. Li, W. Yuan, B. Li, L. Tian, F. Liu and R. Hu, Chem. Commun., 2011, 47, 6858–6860 RSC.
- S. Ding, J. S. Chen, D. Luan, F. Y. C. Boey, S. Madhavi and X. W. D. Lou, Chem. Commun., 2011, 47, 5780–5782 RSC.
- M. Ganji, H. Yazdani and A. Mirnejad, Phys. E, 2010, 42, 2184–2189 CrossRef CAS.
- D. Pantarotto, R. Singh, D. McCarthy, M. Erhardt, J. P. Briand, M. Prato, K. Kostarelos and A. Bianco, Angew. Chem., 2004, 116, 5354–5358 CrossRef.
- S. Meng, P. Maragakis, C. Papaloukas and E. Kaxiras, Nano Lett., 2007, 7, 45–50 CrossRef CAS PubMed.
- I. Willner and B. Willner, Nano Lett., 2010, 10, 3805–3815 CrossRef CAS PubMed.
- J. R. Siqueira, L. Caseli, F. N. Crespilho, V. Zucolotto and O. N. Oliveira, Biosens. Bioelectron., 2010, 25, 1254–1263 CrossRef CAS PubMed.
- C. Chung, Y.-K. Kim, D. Shin, S.-R. Ryoo, B. H. Hong and D.-H. Min, Acc. Chem. Res., 2013, 46, 2211–2224 CrossRef CAS PubMed.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- A. A. Balandin, Nat. Mater., 2011, 10, 569–581 CrossRef CAS PubMed.
- A. C. Neto, F. Guinea, N. Peres, K. S. Novoselov and A. K. Geim, Rev. Mod. Phys., 2009, 81, 109 CrossRef.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924 CrossRef CAS PubMed.
- M. J. Allen, V. C. Tung and R. B. Kaner, Chem. Rev., 2009, 110, 132–145 CrossRef PubMed.
- G. Giovannetti, P. Khomyakov, G. Brocks, V. Karpan, J. van den Brink and P. Kelly, Phys. Rev. Lett., 2008, 101, 026803 CrossRef CAS PubMed.
- C.-W. Lam, J. T. James, R. McCluskey, S. Arepalli and R. L. Hunter, Crit. Rev. Toxicol., 2006, 36, 189–217 CrossRef CAS PubMed.
- C.-W. Lam, J. T. James, R. McCluskey and R. L. Hunter, Toxicol. Sci., 2004, 77, 126–134 CrossRef CAS PubMed.
- J. Muller, F. Huaux, N. Moreau, P. Misson, J.-F. Heilier, M. Delos, M. Arras, A. Fonseca, J. B. Nagy and D. Lison, Toxicol. Appl. Pharmacol., 2005, 207, 221–231 CrossRef CAS PubMed.
- K. Bradley, M. Briman, A. Star and G. Grüner, Nano Lett., 2004, 4, 253–256 CrossRef CAS.
- K. C. Kemp, H. Seema, M. Saleh, N. H. Le, K. Mahesh, V. Chandra and K. S. Kim, Nanoscale, 2013, 5, 3149–3171 RSC.
- L. Yan, Y. B. Zheng, F. Zhao, S. Li, X. Gao, B. Xu, P. S. Weiss and Y. Zhao, Chem. Soc. Rev., 2012, 41, 97–114 RSC.
- K. Gao, G. Chen and D. Wu, PCCP Phys. Chem. Chem. Phys., 2014, 16, 17988–17997 RSC.
- J. M. Anderson, A. Rodriguez and D. T. Chang, Semin. Immunol., 2008, 20, 86–100 CrossRef CAS PubMed.
- M. Ganji, Diamond Relat. Mater., 2009, 18, 662–668 CrossRef CAS.
- S. Gowtham, R. H. Scheicher, R. Ahuja, R. Pandey and S. P. Karna, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 033401 CrossRef.
- S. Gowtham, R. H. Scheicher, R. Pandey, S. P. Karna and R. Ahuja, Nanotechnology, 2008, 19, 125701 CrossRef CAS PubMed.
- H. Vovusha and B. Sanyal, RSC Adv., 2015, 5, 67427–67434 RSC.
- H. K. T. Ozaki, J. Yu, M. J. Han, N. Kobayashi, M. Ohfuti, F. Ishii, T. Ohwaki and H. Weng, available: http://www.openmx-square.org/.
- I. Morrison, D. M. Bylander and L. Kleinman, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 6728 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- A. Banerjee, N. Adams, J. Simons and R. Shepard, J. Phys. Chem., 1985, 89, 52–57 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188 CrossRef.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- S. F. Boys and F. D. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- M. D. Ganji, M. Tajbakhsh and M. Laffafchy, Solid State Sci., 2010, 12, 1547–1553 CrossRef CAS.
- M. Rezvani, M. D. Ganji and M. Faghihnasiri, Phys. E, 2013, 52, 27–33 CrossRef CAS.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS.
- H. Vovusha, S. Sanyal and B. Sanyal, J. Phys. Chem., 2013, 4, 3710–3718 CAS.
- Y. Okamoto, Chem. Phys. Lett., 2006, 420, 382–386 CrossRef CAS.
- M. D. Ganji, S. Hosseini-Khah and Z. Amini-Tabar, PCCP Phys. Chem. Chem. Phys., 2015, 17, 2504–2511 RSC.
- D. Mollenhauer, C. Brieger, E. Voloshina and B. Paulus, J. Phys. Chem. C, 2015, 119, 1898–1904 CAS.
- C. Rajesh, C. Majumder, H. Mizuseki and Y. Kawazoe, J. Chem. Phys., 2009, 130, 124911 CrossRef PubMed.
- M. Ganji and A. Bakhshandeh, Phys. B, 2011, 406, 4453–4459 CrossRef CAS.
- M. D. Ganji, H. Alinezhad, E. Soleymani and M. Tajbakhsh, Phys. E, 2015, 67, 105–111 CrossRef.
- Z. Izakmehri, M. Ardjmand, M. D. Ganji, E. Babanezhad and A. Heydarinasab, RSC Adv., 2015, 5, 48124–48132 RSC.
- K. A. Mkhoyan, A. W. Contryman, J. Silcox, D. A. Stewart, G. Eda, C. Mattevi, S. Miller and M. Chhowalla, Nano Lett., 2009, 9, 1058–1063 CrossRef CAS PubMed.
- D. Li and R. B. Kaner, Nat. Nanotechnol., 2008, 3, 101 CrossRef CAS PubMed.
- S. Liu, M. Hu, T. H. Zeng, R. Wu, R. Jiang, J. Wei, L. Wang, J. Kong and Y. Chen, Langmuir, 2012, 28, 12364–12372 CrossRef CAS PubMed.
- Z.-D. Huang, B. Zhang, S.-W. Oh, Q.-B. Zheng, X.-Y. Lin, N. Yousefi and J.-K. Kim, J. Mater. Chem., 2012, 22, 3591–3599 RSC.
- M. Luo, X. Chen, G. Zhou, X. Xiang, L. Chen, X. Ji and Z. He, Chem. Commun., 2012, 48, 1126–1128 RSC.
- X. Sun, Z. Liu, K. Welsher, J. T. Robinson, A. Goodwin, S. Zaric and H. Dai, Nano Res., 2008, 1, 203–212 CrossRef CAS PubMed.
- Z. Liu, J. T. Robinson, X. Sun and H. Dai, J. Am. Chem. Soc., 2008, 130, 10876–10877 CrossRef CAS PubMed.
- A. Ishii, M. Yamamoto, H. Asano and K. Fujiwara, J. Phys.: Conf. Ser., 2008, 100, 052087 CrossRef.
- K. Nakada and A. Ishii, Solid State Commun., 2011, 151, 13–16 CrossRef CAS.
- M. D. Ganji, N. Sharifi, M. Ardjmand and M. G. Ahangari, Appl. Surf. Sci., 2012, 261, 697–704 CrossRef CAS.
- K. T. Chan, J. Neaton and M. L. Cohen, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 235430 CrossRef.
- I. Tuñón, E. Silla and M. F. Ruiz-López, Chem. Phys. Lett., 2000, 321, 433–437 CrossRef.
- J. H. Jensen and M. S. Gordon, J. Am. Chem. Soc., 1995, 117, 8159–8170 CrossRef CAS.
- F. Floris and J. Tomasi, J. Comput. Chem., 1989, 10, 616–627 CrossRef CAS.
- A. Fernandez-Ramos, E. Cabaleiro-Lago, J. Hermida-Ramon, E. Martınez-Núnez and A. Pena-Gallego, J. Mol. Struct.: THEOCHEM, 2000, 498, 191–200 CrossRef CAS.
- L. Gontrani, B. Mennucci and J. Tomasi, J. Mol. Struct.: THEOCHEM, 2000, 500, 113–127 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: SPE table for defect/A.As complexes, optimized structures and geometrical parameters of A.As and substrates, SPE plot for glycine/graphene configurations, orientation schemes for histidine and phenylalanine molecules approaching the hexagonal network of the graphene, optimized structures and geometrical parameters of graphene/histidine and graphene/phenylalanine complexes, DOS and total density map of GO/A.As complexes, schematic representations of zwitter ionic and neutral glycine in different media. See DOI: 10.1039/c5ra16683g |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.