
Sulfonated nanohydroxyapatite functionalized with 2-aminoethyl dihydrogen phosphate (HAP@AEPH2-SO3H) as a new recyclable and eco-friendly catalyst for rapid one-pot synthesis of 4,4′-(aryl methylene)bis(3-methyl-1H-pyrazol-5-ol)s†
Monireh Zarghani and
Batool Akhlaghinia*
Department of Chemistry, Faculty of Sciences, Ferdowsi University of Mashhad, Mashhad 9177948974, Iran. E-mail: akhlaghinia@um.ac.ir; Fax: +98-51-3879-5457; Tel: +98-51-3880-5527
First published on 30th September 2015
Abstract
Sulfonated nanohydroxyapatite functionalized with 2-aminoethyl dihydrogen phosphate (HAP@AEPH2-SO3H), a new green, recyclable solid acid catalyst, was prepared and characterized using FT-IR, XRD, SEM, TEM and TGA/DTA techniques. The composition of HAP@AEPH2-SO3H was determined as nanohydroxyapatite, while the particles were observed to have nanorod morphology. Size estimations using TEM (10–100 nm) and the crystallinity from XRD (hexagonal phase) are well-matched to the character of nanohydroxyapatite. The catalytic activity of HAP@AEPH2-SO3H was evaluated for the synthesis of 4,4′-(aryl methylene)bis(3-methyl-1H-pyrazol-5-ol)s via one-pot reactions of phenylhydrazine/or hydrazine hydrate, ethyl acetoacetate and aldehydes under solvent-free conditions. This catalyst showed notable advantages, such as environmental friendliness, excellent yields, shorter reaction time, reusability of the inexpensive catalyst and easy workup procedure.
Introduction
4,4′-(Aryl methylene)bis(1H-pyrazol-5-ol)s, an important class of heterocyclic compounds, have exhibited a wide range of biological activities such as antimalarial,1 antifungal,2 anti-inflammatory,3 antimicrobial,4 antinociceptive,5 antiviral,6 and antitumor7 activities. Also, they are used as pesticides,8 fungicides,9 analgesics,10 important intermediates in organic synthesis,11 bis-Schiff bases12 and ligands.13 The conventional chemical approach to 4,4′-(aryl methylene)bis(1H-pyrazol-5-ol)s derivatives is the reaction of initially formed 3-methyl-5-pyrazolone (from the condensation of hydrazine/aryl hydrazine and ethyl acetoacetate) with aldehydes under a variety of reaction conditions. Xanthan sulfuric acid,14 phosphomolybdic acid,15 silica sulfuric acid,16 3-aminopropylated silica gel,17 sodium dodecyl sulfate,18 tetramethyl-tetra-3,4-pyridinoporphyrazinato copper(II) methyl sulfate,19 poly(ethylene glycol)-bound sulfonic acid,20 and others21–32 can be used as catalysts for this transformation. Catalyst free33 and electrocatalytic procedures34 were also used for this preparation. Other methods of preparation include one-pot reactions in which three or more starting materials react together to form a product where all or most of the starting material atoms exist in the final product.35 In comparison, the one-pot character also provides some advantages over the classical stepwise synthetic routes, such as fewer by-products and lower costs, time, and energy. Recently, because of their simple purification, excellent synthetic efficiency, atom economy, operational simplicity, elimination of overflow steps and environmental friendliness, one-pot reactions have been paid much attention.36 Also, one-pot reactions allow formation of complex molecular architectures from simple precursors via the formation of several bonds in a simple synthetic operation, without the need for isolation of intermediates.37 A few methods have been reported for the synthesis of these compounds by one-pot reactions in the presence of silica-bonded N-propylpiperazine sulfamic acid (SBPPSA),38 pyridine trifluoroacetate,39 N,2-dibromo-6-chloro-3,4-dihydro-2H benzo[e][1,2,4]thiadiazine-7-sulfonamide 1,1-dioxide (DCDBTSD),40 ultrasonic irradiation,41 sulfonated rice husk ash (RHA-SO3H)42 and 2-hydroxyethylammonium propionate (2-HEAP).43In recent years, to minimize the use and production of hazardous materials, green chemistry has encouraged chemists to design chemical procedures by using environmentally benign reagents that reduce and prohibit the pollution of nature and ensure perpetual life on the earth.44 Because of the usefulness of one-pot reactions for the synthesis of complex molecules such as drug-like molecules, these kinds of reactions play an important role in combinatorial chemistry and industrial chemistry.45,46
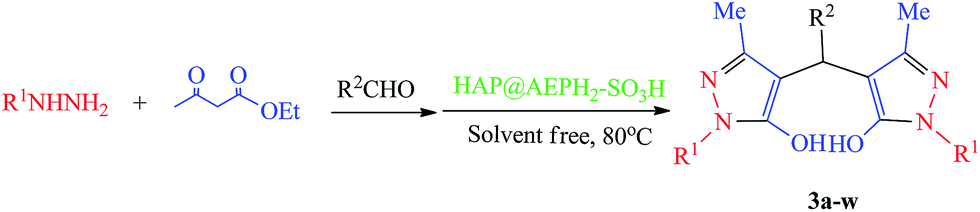
Over the past decades, to overcome some problems of homogeneous catalytic processes, heterogeneous catalysts, such as mixed metal oxides, zeolites, hydrotalcites, solid-supported catalysts, resins, etc., have been particularly attractive, as they possess advantages such as ease of separation of the product, reusability of the catalyst and improved efficiency. On the other hand, heterogeneous catalysts, which have been known for many years, have become strategically vital for efficient and ecofriendly organic transformations. Among the various heterogeneous catalysts, nanohydroxyapatite (nanoHAP [Ca10(PO4)6(OH)2]), an important inorganic nanomaterial with high surface area and low particle size, provides high biocompatibility, great catalytic activity and good adsorption capability. It has been reported that the functional groups, pH, and charge on the surface of nanoHAP have great effects on its surface properties.47 Therefore, to improve the practical applications of nanoHAP as a catalyst in organic synthesis, the modification of the surface of nanoHAP with functional groups is an effective method to obtain unique nanoHAP properties. The use of organophosphates, mostly alkylphosphates, for surface modification of nanoHAP has been described previously.48 It has been found that there are strong interactions between the phosphonate groups of organophosphates with P–OH and perhaps Ca–OH groups on the surface of nanoHAP, which leads to the formation of PS–O–P and P–O–Ca2+ bonds.48c 2-Aminoethyl dihydrogen phosphate (AEPH2), a fairly unknown phospholipid and biodegradable compound, can be used as a surface modification agent. AEPH2 is a bifunctional organic molecule that has both a phosphate group and an amino group.49 Therefore, the phosphate group can be used for binding to the nanoHAP surface and the amino group can also work as an anchoring point for the bonding of several metal ions, and other functional groups. In continuing our efforts toward the development of efficient and environmentally benign heterogeneous catalysts,50 herein, we designed nanoHAP that is modified with AEPH2 as a ligand and then sulfonated with chlorosulfonic acid (HAP@AEPH2-SO3H) (Scheme 1). The catalytic activity of HAP@AEPH2-SO3H as an eco-friendly, reusable heterogeneous catalyst was proved for the synthesis of 4,4′-(aryl methylene)bis(3-methyl-1H-pyrazol-5-ol)s via a one-pot reaction (see Scheme 2).
 | ||
| Scheme 1 Preparation steps of HAP@AEPH2-SO3H. | ||
 | ||
| Scheme 2 One-pot synthesis of 4,4′-(aryl methylene)bis(3-methyl-1H-pyrazol-5-ol)s catalyzed by HAP@AEPH2-SO3H. | ||
Results and discussion
Characterization of catalyst
According to the pathway shown in Scheme 1, HAP@AEPH2-SO3H was synthesized as a novel strong and stable solid acid catalyst. The structure and morphology of the prepared catalyst were characterized by Fourier transform infrared spectroscopy (FT-IR), thermogravimetric analysis (TGA), differential thermal analysis (DTA), scanning electron microscopy (SEM), elemental analysis (CHN), transmission electron microscopy (TEM) and X-ray powder diffraction (XRD), which confirmed the successful preparation of the new catalyst.In order to confirm the successful functionalization of nanoHAP with AEPH2 and then with chlorosulfuric acid, FT-IR spectra of HAP@AEPH2 and HAP@AEPH2-SO3H were recorded (Fig. 1). Fig. 1a depicts the FT-IR spectrum of the nanoHAP functionalized with AEPH2. As can be seen, the stretching and bending vibrations of the OH− ion in hydroxyapatite were detected around 3572 and 630 cm−1, respectively. Also, the broad band at 3450–3050 cm−1 (due to stretching vibration) and the weak band at 1637–1630 cm−1 (due to bending vibration) were attributed to the crystal water and surface adsorbed water. Additional vibrational modes at 1098, 1031, 962, 601, 571, and 471 cm−1 could be ascribed to the asymmetric and symmetric stretching and bending vibrations of the PO43− ions.51 In addition to the principal absorption bands of nanoHAP, the appearance of two absorption bands at 2917 and 2851 cm−1 due to asymmetric and symmetric vibrational frequencies of –CH2 confirmed the successful attachment of AEPH2 on the surface of the nanoHAP (Fig. 1a).52 In the FT-IR spectrum of HAP@AEPH2-SO3H (Fig. 1b), the intensity of the absorption band at 3450–3050 cm−1 was increased because of the presence of more OH groups which appeared after sulfonation. Furthermore, as compared with Fig. 1a, we can see some new absorption bands at 1325 and 1155 cm−1, which are attributed to the asymmetric and symmetric stretching frequencies of S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O.53 These absorption bands reveal that the –SO3H group has been successfully grafted onto the surface of HAP@AEPH2.
O.53 These absorption bands reveal that the –SO3H group has been successfully grafted onto the surface of HAP@AEPH2.
 | ||
| Fig. 1 FT-IR spectrum of (a) HAP@AEPH2 and (b) HAP@AEPH2-SO3H. | ||
The crystallinity and phase identification of HAP@AEPH2 and HAP@AEPH2-SO3H were determined by using X-ray diffraction (XRD). As presented in Fig. 2, characteristic peaks with strong intensities appeared at angles corresponding to the (002), (211), (300), (202), (310), (222), (213) and (004) crystallographic faces, which are in agreement with the standard data of nanoHAP (JCPDS: 74-0565).54 The XRD results also demonstrate the presence of hexagonal phase nanoHAP with high crystallinity.
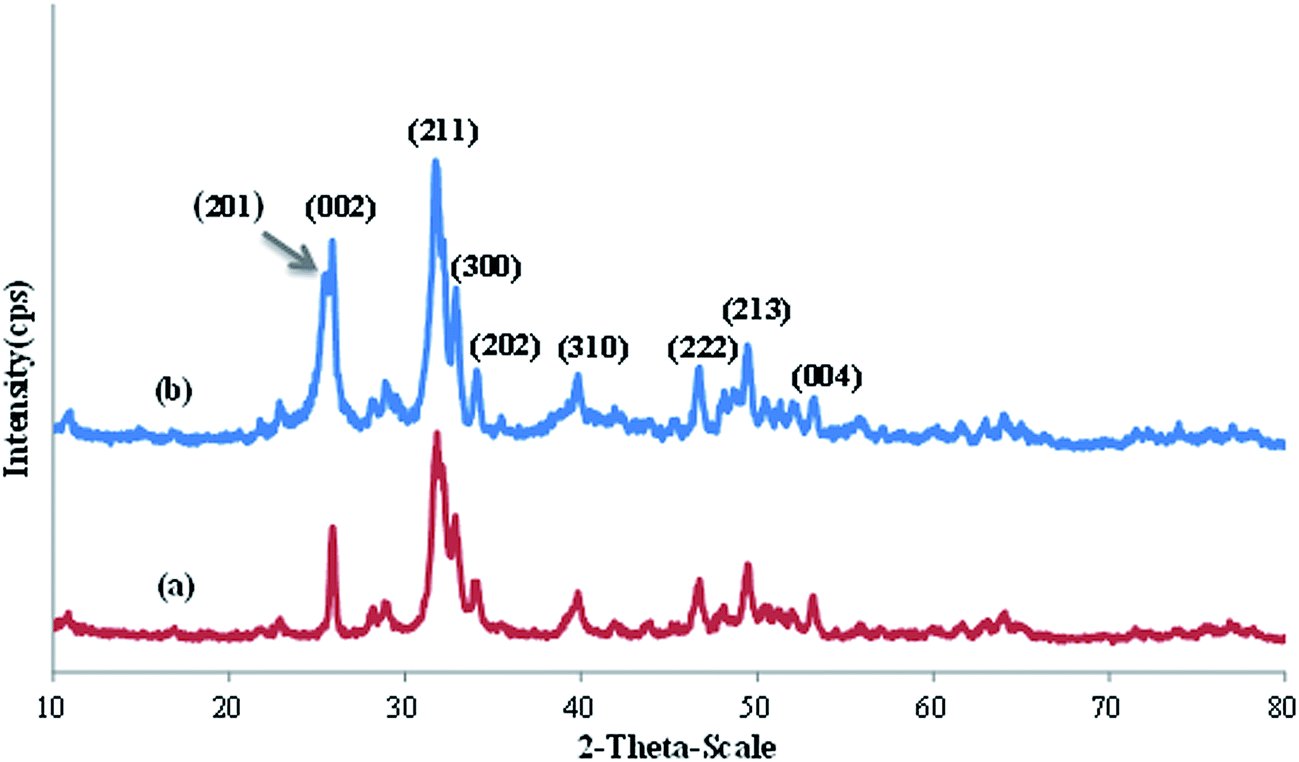 | ||
| Fig. 2 The XRD patterns of (a) HAP@AEPH2 and (b) HAP@AEPH2-SO3H. | ||
Although XRD analysis of HAP@AEPH2-SO3H showed no considerable broadening or shifting of peaks when compared with the XRD of HAP@AEPH2 (Fig. 2), an additional peak observed in the HAP@AEPH2-SO3H pattern at 2θ = 25.4° could be attributed to a small amount of CaSO4 impurity due to sulfonation (JCPDS: 37-1496) (Fig. 2b).55
In spite of this, it could be seen that the modification of nanoHAP with AEPH2 and chlorosulfuric acid did not change the crystal structure of the nanoHAP.
In addition, scanning electron microscopy and transmission electron microscopy (SEM and TEM) images were used to investigate the morphology and size of the HAP@AEPH2-SO3H nanoparticles. Fig. 3a and b represent the SEM micrographs of the HAP@AEPH2-SO3H nanoparticles. These images demonstrate that the HAP@AEPH2-SO3H nanoparticles have a uniform nanorod morphology which is similar to the particle morphologies reported elsewhere.56 The TEM images of the HAP@AEPH2-SO3H nanoparticles shown in Fig. 3c and d revealed that the synthesized nanoparticles are rod-like in shape with a mean size range of 10–100 nm. They also indicate that in comparison with the literature57 there is no change in the shape of nanoHAP after being functionalized with AEPH2 and sulfonic acid and the surface morphology is also retained.
 | ||
| Fig. 3 (a and b) SEM images of HAP@AEPH2-SO3H, (c and d) TEM images of HAP@AEPH2-SO3H. | ||
Quantitative determination of the organic functional groups loaded on the surface of nanoHAP was carried out using TGA and DTA (Fig. 4). For HAP@AEPH2, weight loss at temperatures below 200 °C (4.00%) is probably related to the physically adsorbed water on the catalyst surface. Also, the weight loss from 200 to 500 °C is associated with the decomposition of AEPH2 groups grafted to the nanoHAP surface (Fig. 4a).58 The TGA curve of HAP@AEPH2-SO3H (Fig. 4b) shows three weight loss steps. The initial weight loss up to 200 °C is due to absorbed water and the second step involves the decomposition of AEPH2, which started after 200 °C and continued up to 500 °C. The last step can be observed between 500 °C and 850 °C, corresponding to the thermal decomposition of SO3H groups embedded via AEPH2 on the surface of nanoHAP.59
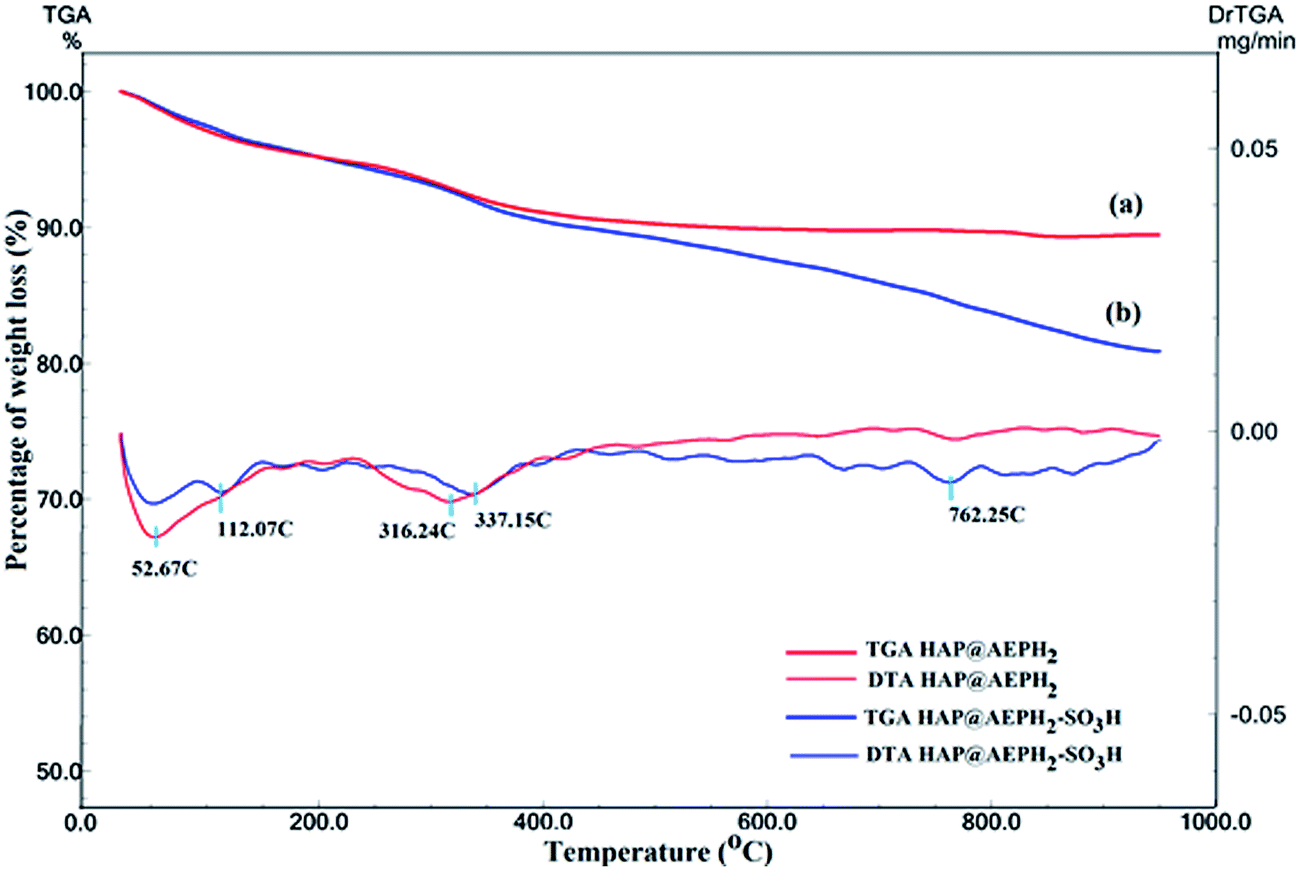 | ||
| Fig. 4 The TGA/DTA thermograms of (a) HAP@AEPH2 and (b) HAP@AEPH2-SO3H. | ||
According to the TGA curves, it can be estimated that the amounts of AEPH2 and sulfonic acid functionalized onto the surface of the catalyst are about 5.60 and 8.00%, respectively.
In addition to the TGA/DTA analysis, the contents of AEPH2 and sulfonic acid were also determined by elemental analysis (Table 1). As can be seen in Table 1, the elemental analysis of the catalyst indicated that 1.008 mmol of AEPH2 and 1.040 mmol of sulfonic acid are incorporated into 1.000 g of HAP@AEPH2-SO3H, which is in good agreement with the data obtained from TGA.
| Samples | TGA (%) | Elemental analysis (w/w%) | ||||
|---|---|---|---|---|---|---|
| H2O | AEPH2 | SO3H | C | N | S | |
| HAP@AEPH2 | 4.000 (2.200 mmol g−1) | 5.600 (1.010 mmol g−1) | — | 2.409 (1.004 mmol g−1) | 1.397 (1.000 mmol g−1) | — |
| HAP@AEPH2-SO3H | 4.000 (2.200 mmol g−1) | 5.600 (1.010 mmol g−1) | 8.000 (1.065 mmol g−1) | 2.419 (1.008 mmol g−1) | 1.409 (1.007 mmol g−1) | 3.32 (1.040 mmol g−1) |
Moreover, the number of acidic sites in HAP@AEPH2-SO3H was determined by back-titration analysis of the catalyst: 100 mg of catalyst was added to a solution of NaOH (15 mL, 0.1 N). The resulting suspension was maintained at room temperature overnight under stirring. After that, the suspension was filtered. The filtrate was neutralized by a standard solution of HCl (0.1 M). The consumed volume of HCl (14.1 mL) determined the amount of loaded NHSO3H per 1.000 g of HAP@AEPH2-SO3H (0.900 mmol of NHSO3H per 1.000 g of catalyst). This result is in good agreement with those obtained from TGA and elemental analysis.
Catalytic synthesis of 4,4′-(aryl methylene)bis(3-methyl-1H-pyrazol-5-ol)
In continuation of our studies to develop new catalysts for organic transformations50 and to replace the conventional, toxic and polluting Brønsted acid catalysts with eco-friendly, reusable, heterogeneous catalysts, HAP@AEPH2-SO3H was used as a solid acid catalyst in the one-pot preparation of 4,4′-(aryl methylene)bis(3-methyl-1H-pyrazol-5-ol)s from phenylhydrazine or hydrazine hydrate, ethyl acetoacetate and aldehydes (Scheme 2).To find an appropriate reaction medium for the synthesis of 4,4′-(aryl methylene)bis(3-methyl-1H-pyrazol-5-ol)s in the presence of a catalytic amount of HAP@AEPH2-SO3H, the one pot reaction between phenylhydrazine (2 mmol), ethyl acetoacetate (2 mmol) and benzaldehyde (1 mmol) was selected as a model reaction. The model reaction was examined under different reaction conditions, such as different solvents, different temperatures and various amounts of catalyst (Table 2). A blank experiment without catalyst under solvent-free conditions at 80 °C gave very low yield (10%) after 24 h (Table 2, entry 1). The efficient catalytic activity of HAP@AEPH2-SO3H was confirmed by performing the model reaction in the presence of nanoHAP and nanoHAP-NH2. The desired product was scarcely obtained (20%) in the presence of nanoHAP and nano HAP-NH2 under solvent-free conditions at 80 °C (Table 2, entries 2–3). Applying 2 mol% of HAP@AEPH2-SO3H (contains 0.02 mmol of acid) under solvent-free conditions at 80 °C produced 4,4′-(phenylmethylene)bis(3-methyl-1H-pyrazol-5-ol) in high yield after 2 (min) (Table 2, entry 4). We examined the effect of different temperatures on the model reaction under solvent free conditions. The reactions took a long time to achieve a high yield of the desired product (Table 2, entries 5–8). To evaluate the effect of different amounts of HAP@AEPH2-SO3H, the model reaction was performed in the presence of 1 mol% (contains 0.01 mmol of acid) and 1.5 mol% (contains 0.015 mmol of acid) of catalyst (Table 2, entries 9–10). The best result was obtained by applying 1.5 mol% of catalyst under solvent free conditions at 80 °C. To examine the effect of solvent, various solvents were screened (H2O, EtOH, H2O/EtOH (1/1) and CH3CN) in the preparation of 4,4′-(phenylmethylene)bis(3-methyl-1H-pyrazol-5-ol) in the presence of 1.5 mol% of HAP@AEPH2-SO3H (Table 2, entries 11–14). It is observed that solvent-free conditions gave the best result for this transformation.
| Entry | Catalyst (mol%) | Solvent | Temperature (°C) | Time (min) | Isolated yield (%) |
|---|---|---|---|---|---|
| a The reaction was performed in the presence of nanoHAP.b The reaction was performed in the presence of nanoHAP-NH2. | |||||
| 1 | — | Solvent-free | 80 | 24 (h) | 10 |
| 2a | 2 | Solvent-free | 80 | 3 (h) | 20 |
| 3b | 2 | Solvent-free | 80 | 3 (h) | 20 |
| 4 | 2 | Solvent-free | 80 | 2 | 98 |
| 5 | 2 | Solvent-free | 70 | 6 | 92 |
| 6 | 2 | Solvent free | 60 | 10 | 94 |
| 7 | 2 | Solvent-free | 40 | 45 | 95 |
| 8 | 2 | Solvent-free | r.t. | 2 (h) | 45 |
| 9 | 1 | Solvent-free | 80 | 10 | 95 |
| 10 | 1.5 | Solvent-free | 80 | 3 | 98 |
| 11 | 1.5 | H2O | 80 | 10 | 75 |
| 12 | 1.5 | EtOH | 80 | 15 | 70 |
| 13 | 1.5 | H2O/EtOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
80 | 15 | 65 |
| 14 | 1.5 | CH3CN | 80 | 10 | 75 |
With this result in hand, in the next step, to explore the scope and efficiency of this one-pot reaction we examined a range of various aromatic aldehydes under the optimized reaction conditions. For this purpose, phenyl hydrazine or hydrazine (2 mmol), ethyl acetoacetate (2 mmol) and a broad range of structurally diverse aromatic aldehydes (1 mmol) were condensed in the presence of 1.5 mol% of HAP@AEPH2-SO3H to achieve the desired product. The results are summarized in Table 3. Aromatic aldehydes (bearing electron withdrawing and electron-donating groups) led to rapid formation of products 3a–q (Table 3, entries 1–17) in high yields. Also, heteroaromatic aldehydes such as pyridine carboxaldehydes, and allylic aldehydes such as cinnamaldehyde, led to products 3r (94%), 3s (93%), and 3t (90%), respectively (Table 3, entries 18–20).
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | Product | Time (min) | Isolated yield (%) |
| 1 | C6H5 | C6H5 | 3a | 3 | 98 |
| 2 | C6H5 | 4-OHC6H4 | 3b | 2 | 95 |
| 3 | C6H5 | 3-OHC6H4 | 3c | 3 | 93 |
| 4 | C6H5 | 2-OHC6H4 | 3d | 4 | 90 |
| 5 | C6H5 | 4-OMeC6H4 | 3e | 3 | 98 |
| 6 | C6H5 | 3-OMeC6H4 | 3f | 7 | 93 |
| 7 | C6H5 | 4-MeC6H4 | 3g | 5 | 93 |
| 8 | C6H5 | 3-MeC6H4 | 3h | 7 | 85 |
| 9 | C6H5 | 5-Br-2-OHC6H3 | 3i | 7 | 92 |
| 10 | C6H5 | 4-iPrC6H4 | 3j | 5 | 80 |
| 11 | C6H5 | 2-ClC6H4 | 3k | 7 | 80 |
| 12 | C6H5 | 4-ClC6H4 | 3l | 3 | 98 |
| 13 | C6H5 | 2,4-(Cl)2C6H3 | 3m | 7 | 85 |
| 14 | C6H5 | 4-BrC6H4 | 3n | 2 | 97 |
| 15 | C6H5 | 3-NO2C6H4 | 3o | 3 | 80 |
| 16 | C6H5 | 2-NO2C6H4 | 3p | 5 | 80 |
| 17 | C6H5 | 1-Naphthyl | 3q | 5 | 95 |
| 18 | C6H5 | 2-Pyridyl | 3r | 2 | 94 |
| 19 | C6H5 | 3-Pyridyl | 3s | 3 | 93 |
| 20 | C6H5 | C6H5CHCH2 |
3t | 5 | 90 |
| 21 | H | C6H5 | 3u | 10 | 98 |
| 22 | H | 4-ClC6H4 | 3v | 8 | 98 |
| 23 | H | 4-MeC6H4 | 3w | 10 | 95 |
We also used hydrazine instead of phenylhydrazine in this reaction (Table 3, entries 21–23). The desired products were also obtained rapidly in excellent yields.
The structures of all synthesized compounds 3a–w have been established by their melting points, FT-IR spectroscopy and 1H NMR spectroscopy. Moreover, the selected compounds were further identified by 13C NMR spectroscopy, mass spectrometry and elemental analysis.
The FT-IR spectrum of the purified products displayed characteristic signals for OH, CN and CC bonds around 3450–2500, 1600 and 1499 cm−1, respectively. Also, the 1H NMR spectra exhibited a sharp singlet resonating at around 5.50–4.89 ppm due to the aryl methylene proton and two broad peaks around 13.91 and 12.40 ppm revealed two –OH groups. In the 13C NMR spectra, a signal at 32 ppm is assigned to the aryl methylene carbon. The elemental analysis of the synthesized compounds was also in good conformity with the proposed structures.
To further elucidate the structure of the products, mass spectrometry was provided. As can be seen, the molecular ion was not evident in some of the mass spectra because of the very facile cleavage of C–C bond between the aryl methylene carbon and the pyrazole ring. However, some useful fragmentation information for each compound has been described, which can be beneficial for depicting the desired products.
A reasonable mechanism for the formation of the 4,4′-(aryl methylene)bis(1H-pyrazol-5-ol)s derivatives is proposed in Scheme 3. The reaction involves the initial formation of pyrazolone (VI) (which is in equilibrium with its other tautomeric form (VII)) by the reaction between the protonated form of ethyl acetoacetate (I) and phenylhydrazine or hydrazine. Afterwards, in acidic media, condensation of the intermediate (VII) with aldehyde and subsequent dehydration, leads to the formation of IX. A Michael addition reaction between IX and VII produced two tautomeric forms XI and XII followed by release of the acidic catalyst. Then, the solid acid catalyst re-enters the catalytic cycle.
 | ||
| Scheme 3 Proposed mechanism for the synthesis of 4,4′-(aryl methylene)bis(3-methyl-1H-pyrazol-5-ol)s using solid acid catalyst (HAP@AEPH2-SO3H). | ||
Among the various reactions, we found that in the reaction between 4-Me2N–C6H4–CHO and 3-methyl-1-phenyl-1H-pyrazol-5-ol (VII), no desired product was obtained. Instead, (E)-4-(4-(dimethylamino)benzylidene)-3-methyl-1-phenyl-1H-pyrazol-5(4H)-one was formed as the product, which was established by FT-IR, mass spectroscopy and 1H NMR spectroscopy (see ESI†) (Scheme 3). This effect can be explained by a conjugative effect that is responsible for greater stability as compared to its analogues (formed by use of other aldehydes).60
In another study, the reusability of the catalyst, which is one of the most important benefits and makes it useful for commercial applications, was examined for the model reaction. In this experiment, after completion of the reaction, the reaction mixture was boiled in EtOAc and then centrifuged to separate the catalyst. To remove all organic compounds, the catalyst was refluxed in EtOAc (3 × 10) and centrifuged consecutively. The recycled catalyst was dried at 50 °C under vacuum for 3 h and used in the next run of the model reaction. The results of this experiment and five subsequent experiments were almost consistent in yields (97, 97, 94, 92, 85%). Although slightly more time was required to complete the reaction in the fifth run, the yields are comparable to those seen earlier (Fig. 5). Elemental analysis of the reused catalyst (C 2.398%, N 1.368%, S 3.101%) showed that there was no significant leaching of the ligand AEPH2 from the catalyst to the reaction mixture, but a small amount of leaching of SO3H can be observed away from the surface of the catalyst after five runs of the reaction.
 | ||
| Fig. 5 Synthesis of 4,4′-(phenylmethylene)bis(3-methyl-1H-pyrazol-5-ol) in the presence of reused HAP@AEPH2-SO3H. *The data referred to conversion of benzaldehyde. | ||
We compared the efficiency of HAP@AEPH2-SO3H with previously reported catalysts in the literature that were applied in the one-pot preparation of 4,4′-(aryl methylene)bis(1H-pyrazol-5-ol)s derivatives. The results are shown in Table 4. It is clear from Table 4 that HAP@AEPH2-SO3H promoted the reaction more effectively than a number of other catalysts, particularly in terms of the reaction yield and also the time required to complete the reaction.
| Entry | Catalyst | Solvent | Temperature (°C) | Time (min) | Yield (%) | Reference |
|---|---|---|---|---|---|---|
| a 2-Hydroxy ethylammonium propionate (2-HEAP).b Silica-bonded N-propylpiperazine sulfamic acid (SBPPSA).c N,2-Dibromo-6-chloro-3,4-dihydro-2H benzo[e][1,2,4]thiadiazine-7-sulfonamide 1,1-dioxide (DCDBTSD). | ||||||
| 1 | Ultrasonic irradiation | H2O/EtOH(1:1) |
r.t. | 15 | 98 | 45 |
| 2 | 2-HEAPa | Solvent-free | 90 | 30 | 91 | 47 |
| 3 | SBPPSAb | Solvent-free | 80 | 45 | 93 | 42 |
| 4 | RHA-SO3H | Solvent-free | 80 | 3 | 91 | 46 |
| 5 | DCDBTSDc | Solvent-free | 80 | 100 | 74 | 44 |
| 6 | HAP@AEPH2-SO3H | Solvent-free | 80 | 3 | 98 | Present study |
Experimental
General
The purity determinations of the products were accomplished by TLC on silica gel polygram STL G/UV 254 plates. The melting points of the products were determined with an Electrothermal Type 9100 melting point apparatus. The FTIR spectra were recorded on pressed KBr pellets using an AVATAR 370 FT-IR spectrometer (Therma Nicolet spectrometer, USA) at room temperature in the range between 4000 and 400 cm−1 with a resolution of 4 cm−1, and each spectrum was the average of 32 scans. NMR spectra were recorded on a NMR Bruker Avance spectrometer at 400 and 300 MHz in DMSO-d6 as the solvent in the presence of tetramethylsilane as the internal standard. Elemental analysis was performed using a Thermo Finnigan Flash EA 1112 Series instrument (furnace: 900 °C, oven: 65 °C, flow carrier: 140 mL min−1, flow reference: 100 mL min−1). Mass spectra were recorded with a CH7A Varianmat Bremen instrument at 70 eV electron impact ionization, in m/z (rel%). TGA and DTA analysis were performed using a Shimadzu Thermogavimetric Analyzer (TG-50) in the temperature range of 25–900 °C at a heating rate of 10 °C min−1 under air atmosphere. SEM images were recorded using a Leo 1450 VP scanning electron microscope operating at an acceleration voltage of 20 kV and resolution of about 2 nm, and imaging with a magnification of 20 to 300000 times (LEO, Germany). Transmission electron microscopy (TEM) was performed with a Leo 912 AB microscope (Zeiss, Germany) with an accelerating voltage of 120 kV, imaging with a magnification of 80 to 500000 times, and equipped with a high resolution CCD Camera. The crystal structure of the catalyst was analyzed by XRD using a D8 ADVANCE-Bruker diffractometer operated at 40 kV and 30 mA, utilizing CuKα radiation (λ = 0.154 Å), at a step size of 0.040° and step time of 1.5 s. The diffraction angles (2θ) were scanned from 10° to 80°. All yields refer to isolated products after purification by recrystallization.
Preparation of nanoHAP functionalized with AEPH2 (HAP@AEPH2)
To a solution of Ca(NO3)2·4H2O (50 mL, 1.08 M, at pH adjusted to 10 with NH4OH), in a three-necked 500 mL round-bottomed flask equipped with a condenser, argon gas inlet tube and dropping funnel, a solution of (NH4)2HPO4 (50 mL, 0.65 M at pH adjusted to 10 with NH4OH) was added at 90 °C with stirring. AEPH2 (0.8 g, 5 mmol) was added to the resulting suspension. The precipitate was maintained in contact with the reaction solution for 5 h at 90 °C under stirring. Then, the suspension was centrifuged at 10000 rpm for 10 min and repeatedly washed with CO2-free distilled water (3 × 20 mL). The product (nanoHAP functionalized with AEPH2) was dried at 50 °C overnight.48d
Preparation of HAP@AEPH2-SO3H
To a magnetically stirred mixture of HAP@AEPH2 (1 g) in 20 mL of dry dichloromethane, 0.5 mL (0.75 mmol) of chlorosulfuric acid was added drop by drop at room temperature. After 20 min the suspension was filtered and washed with 20 mL of dichloromethane and dried at room temperature to afford HAP@AEPH2-SO3H as a white powder.Typical procedure for preparation of 4,4′-(phenylmethylene)bis(3-methyl-1 phenyl-1H-pyrazol-5-ol)
Phenylhydrazine (2 mmol, 0.216 g) was added to a mixture of HAP@AEPH2-SO3H (1.5 mol%, 0.015 g) and ethyl acetoacetate (2 mmol, 0.260 g) at 80 °C. The resultant mixture was stirred for 1 min. Then, benzaldehyde (1 mmol, 0.106 g) was added to the reaction mixture at 80 °C. The viscous mixture was stirred vigorously for the appropriate time (5 min). The progress of the reaction was monitored by thin layer chromatography (TLC). After completion of the reaction, the reaction mixture was washed with hot ethanol (2 × 5 mL). The resultant hot suspension was filtered off. The filtrate was cooled and the crude product was precipitated. The crude products were purified by recrystallization from ethanol (0.427 g, 98%). To recycle the catalyst, the precipitate (HAP@AEPH2-SO3H) was boiled in EtOAc (5 mL) for 10 min and then centrifuged at 10000 rpm for 5 min. To remove all organic compounds, the catalyst was refluxed in EtOAc (3 × 10) for 30 min and centrifuged consecutively. The recycled catalyst was dried at 50 °C under vacuum for 3 h and used in the next run of the model reaction.
Spectral data
N),1497, 1449 (CC), 1412, 1368, 1313, 1263, 1127, 1029, 857, 792, 754, 693, 596, 506, 449; 1H NMR (300 MHz, DMSO-d6, ppm) δ 2.33 (s, 6H, 2CH3), 4.98 (s, 1H, CH), 7.18–7.27 (m, 7H, Ph), 7.45 (t, J = 7.8 Hz, 4H, Ph), 7.72 (d, J = 8.1 Hz, 4H, Ph), 13.96 (br, 1H, OH); 13C NMR (75 MHz, DMSO-d6 ppm) δ 12.1, 33.5, 120.9, 126.0, 126.3, 127.6, 128.6, 129.0, 129.4, 137.7, 142.6, 146.8; MS, m/z (%): 259 [M+ − 177], 183 [M+ − 251], 172 [M+ − 264].N), 1580, 1504, 1454 (CC), 1371, 1275, 1245, 1173, 1111, 1029, 867, 816, 779, 749, 690, 583, 498, 453; 1H NMR (300 MHz, DMSO-d6, ppm) δ 2.31 (s, 6H, 2CH3), 4.87 (s, 1H, CH), 6.68 (d, J = 8.7 Hz, 2H, Ph), 7.06 (d, J = 8.4 Hz, 2H, Ph), 7.25 (t, J = 7.2 Hz, 2H, Ph), 7.45 (t, J = 7.8 Hz, 4H, Ph), 7.72 (d, J = 7.8 Hz, 4H, Ph), 9.21 (s, 1H, OH), 13.95 (br, 1H, OH); 13C NMR (75 MHz, DMSO-d6 ppm) δ 19.0, 32.8, 105.7, 115.3, 120.9, 126.0, 128.5, 129.0, 129.3, 132.7, 137.8, 146.6, 155.8, 155.9; anal. calcd for C27H24N4O3: C, 71.67; H, 5.35; N, 12.38, found: C, 70.71; H, 5.8; N, 11.62%; MS, m/z (%): 275 [M+ − 177], 184 [M+ − 268], 173 [M+ − 279].N), 1498 (CC), 1455, 1405, 1368, 1284, 1155, 1029, 996, 754, 692, 503, 412; 1H NMR (300 MHz, DMSO-d6, ppm) δ 2.32 (s, 6H, 2CH3), 4.89 (s, 1H, CH), 6.58 (d, J = 7.8 Hz, 1H, Ph), 6.65–6.70 (m, 2H, Ph), 7.07 (t, J = 7.8 Hz, 1H, Ph), 7.26 (t, J = 7.8 Hz, 2H, Ph), 7.46 (t, J = 7.8 Hz, 4H, Ph), 7.73 (d, J = 7.8 Hz, 4H, Ph), 9.26 (s, 1H, OH), 13.98 (br, 1H, OH); 13C NMR (75 MHz, DMSO-d6 ppm) δ 12.0, 33.4, 113.3, 114.6, 118.3, 118.8, 120.9, 126.0, 129.4, 129.4, 137.5, 138.1, 144.0, 146.8, 157.6.N), 1574, 1499 (CC), 1454, 1367, 1286, 1217, 1119, 1041, 912, 879, 847, 751, 687, 606, 586, 506, 440; 1H NMR (300 MHz, DMSO-d6, ppm) δ 2.29 (s, 6H, 2CH3), 5.18 (s, 1H, CH), 6.69–6.77 (m, 2H, Ph), 6.96–7.02 (m, 1H, Ph), 7.23–7.27 (m, 2H, Ph), 7.44 (t, J = 7.5 Hz, 4H, Ph), 7.55 (d, J = 7.2 Hz, 1H, Ph), 7.70 (d, J = 8.1 Hz, 4H, Ph), 12.34 (br, 1H, OH); MS, m/z (%): 275 [M+ − 177], 256 [M+ − 196], 173 [M+ − 279].N), 1581, 1507, 1459 (CC), 1406, 1373, 1279, 1249, 1180, 1111, 1033, 966, 812, 751, 691, 596, 498, 442; 1H NMR (300 MHz, DMSO-d6, ppm) δ 2.32 (s, 6H, 2CH3), 3.71 (s, 3H, OCH3), 4.91 (s, 1H, CH), 6.84 (d, J = 8.4 Hz, 2H, Ph), 7.15–7.27 (m, 4H, Ph),7.45 (t, J = 7.8 Hz, 4H, Ph), 7.71 (d, J = 7.8 Hz, 4H, Ph), 13.97 (br, 1H, OH); 13C NMR (75 MHz, DMSO-d6, ppm) δ 12.1, 32.8, 55.4, 113.9, 120.9, 126.0, 128.6, 129.3, 134.4, 146.6, 157.9; MS, m/z (%): 288 [M+ − 178], 258 [M+ − 208], 183 [M+ − 283].N), 1581, 1502, 1485 (CC), 1405, 1364, 1311, 1274, 1188, 1129, 1042, 1004, 877, 790, 752, 693, 636, 599, 496, 453; 1H NMR (300 MHz, DMSO-d6, ppm) δ 2.32 (s, 6H, 2CH3), 3.69 (s, 3H, OCH3), 4.93 (s, 1H, CH), 6.76–6.87 (m, 3H, Ph), 7.18–7.28 (m, 3H, Ph), 7.45 (t, J = 7.8 Hz, 4H, Ph), 7.71 (d, J = 7.8 Hz, 4H, Ph), 12.27 (br, 1H, OH), 13.98 (br, 1H, OH); anal. calcd for C28H26N4O3: C, 72.09; H, 5.62; N, 12.01, found: C, 71.9; H, 5.35; N, 11.9%; MS, m/z (%): 289 [M+ − 177], 259 [M+ − 207], 184 [M+ − 282].N), 1580, 1501, 1457 (CC), 1408, 1372, 1291, 1196, 1131, 1070, 1024, 882, 802, 750, 688, 608, 592, 499, 444; 1H NMR (300 MHz, DMSO-d6, ppm) δ 2.25 (s, 3H, PhCH3), 2.33 (s, 6H, 2CH3), 4.93 (s, 1H, CH), 7.08 (d, J = 8.1 Hz, 2H, Ph), 7.16 (d, J = 7.8 Hz, 2H, Ph), 7.25 (t, J = 7.5 Hz, 2H, Ph), 7.45 (t, J = 7.5 Hz, 4H, Ph), 7.72 (d, J = 7.5 Hz, 4H, Ph), 13.94 (br, 1H, OH); 13C NMR (75 MHz, DMSO-d6, ppm) δ 12.1, 21.0, 33.2, 105.2, 120.9, 126.0, 127.5, 129.1, 129.4, 135.3, 137.8, 139.5, 146.7; anal. calcd for C28H26N4O2: C, 74.67; H, 5.82; N, 12.44, found: C, 74.31; H, 5.8; N, 12.24%. MS, m/z (%): 273 [M+ − 177], 259 [M+ − 191], 184 [M+ − 266].N), 1578, 1499 (CC), 1457, 1410, 1372, 1289, 1180, 1102, 1024, 902, 800, 752, 691, 597, 499, 442; 1H NMR (300 MHz, DMSO-d6, ppm) δ 2.25 (s, 3H, PhCH3), 2.32 (s, 6H, 2CH3), 4.93 (s, 1H, CH), 7.00–7.25 (m, 6H, Ph), 7.45 (m, 4H, Ph), 7.72 (d, J = 7.2 Hz, 4H, Ph), 13.95 (br, 1H, OH); 13C NMR (75 MHz, DMSO-d6, ppm) δ 12.1, 21.7, 33.5, 120.9, 124.7, 126.0, 127.1, 128.2, 128.5, 129.4, 137.5, 142.6, 146.7; MS, m/z (%): 272 [M+ − 178], 258 [M+ − 192], 183 [M+ − 267].N), 1501, 1415 (CC), 1371, 1286, 1184, 1025, 792, 753, 690, 600, 498, 449. 1H NMR (300 MHz, DMSO-d6, ppm) δ 1.17 (d, J = 6.6 Hz, 6H, 2CH3), 2.32 (s, 6H, 2CH3), 2.79–2.88 (m, 1H, CH), 4.92 (s, 1H, CH), 7.13–7.28 (m, 6H, Ph), 7.45 (t, J = 7.5 Hz, 4H, Ph), 7.71 (d, J = 7.8 Hz, 4H, Ph), 12.37 (br, 1H, OH), 13.97 (br, 1H, OH); anal. calcd for C30H30N4O2: C, 75.29; H, 6.32; N, 11.71, found: C, 75.1; H, 6.3; N, 11.53%; MS, m/z (%): 301 [M+ − 177], 258 [M+ − 220], 184 [M+ − 294].N), 1540, 1499 (CC), 1458, 1400, 1369, 1305, 1217, 1131, 1082, 1033, 898, 837, 792, 746, 689, 637, 568, 501; 1H NMR (300 MHz, DMSO-d6, ppm) δ 2.30 (s, 6H, 2CH3), 5.15 (s, 1H, CH), 7.24–7.34 (m, 4H, Ph), 7.40–7.47 (m, 5H, Ph), 7.68–7.79 (m, 5H, Ph), 13.97 (br, 1H, OH); 13C NMR (75 MHz, DMSO-d6, ppm) δ 12.3, 32.1, 121.1, 126.1, 127.3, 128.5, 129.4, 129.9, 130.7, 132.3, 137.6, 139.8, 146.5; MS, m/z (%): 472 [M+ + 2], 294 [M+ − 176], 259 [M+ − 211], 184 [M+ − 286].N), 1578, 1501, 1486 (CC), 1408, 1372, 1295, 1204, 1086, 1014, 834, 810, 747, 688, 592, 599, 439; 1H NMR (300 MHz, DMSO-d6, ppm) δ 2.33 (s, 6H, 2CH3), 4.98 (s, 1H, CH), 7.25–7.72 (m, 10H, Ph), 7.71 (d, J = 7.8 Hz, 4H, Ph); 13C NMR (75 MHz, DMSO-d6, ppm) δ 12.0, 32.9, 104.8, 121.0, 126.1, 128.4, 129.4, 129.6, 131.0, 137.0, 137.7, 141.5, 146.7; MS, m/z (%): 470 [M+], 292 [M+ − 178], 259 [M+ − 212].N), 1558, 1498 (CC), 1462, 1405, 1367, 1311, 1217, 1143, 1074, 902, 836, 790, 754, 693, 637, 571, 502; 1H NMR (300 MHz, DMSO-d6, ppm) δ 1.84 (s, 6H, 2CH3), 5.59 (s, 1H, CH), 7.17–7.45 (m, 9H, Ph), 7.73 (d, J = 7.5 Hz, 4H), 10.83 (br, 1H, OH), 11.30 (br, 1H, OH); 13C NMR (75 MHz, DMSO-d6, ppm) δ 11.7, 34.3, 105.4, 118.3, 118.5, 120.1, 124.6, 125.3, 128.6, 129.0, 129.5, 129.9, 136.1, 137.8, 163.5.N), 1578, 1499 (CC), 1456, 1406, 1368, 1306, 1184, 1072, 1026, 1009, 910, 816, 753, 690, 593, 499; 1H NMR (400 MHz, DMSO-d6, ppm) δ 2.29 (s, 6H, 2CH3), 4.93 (s, 1H, CH), 7.16–7.23 (m, 4H, Ph), 7.44–7.45 (m, 6H, Ph), 7.66–7.67 (m, 4H, Ph).N), 1573, 1527, 1501 (CC), 1418, 1349, 1285, 1200, 1106, 1004, 976, 861, 831, 803, 754, 691, 597, 497, 443; 1H NMR (300 MHz, DMSO-d6, ppm) δ 2.37 (s, 6H, 2CH3) 5.17 (s, 1H, CH), 7.26 (t, J = 7.5 Hz, 2H, Ph), 7.46 (t, J = 7.5 Hz, 4H, Ph), 7.60 (t, J = 8.1, 1H, Ph), 7.64–7.76 (m, 5H, Ph), 8.08–8.10 (m, 2H, Ph), 13.86 (br, 1H, OH); MS, m/z (%): 303 [M+ − 178], 257 [M+ − 224], 183 [M+ − 228].N), 1558, 1521, 1498 (CC), 1458, 1406, 1372, 1315, 1164, 1127, 1070, 898, 837, 788, 755, 677, 628, 567, 502; 1H NMR (300 MHz, DMSO-d6, ppm) δ 2.26 (s, 6H, 2CH3), 5.43 (s, 1H, CH), 7.25–7.28 (m, 2H, Ph), 7.42–7.47 (m, 5H, Ph), 7.66–7.74 (m, 7H, Ph), 13.50 (br, 1H, OH); 13C NMR (75 MHz, DMSO-d6, ppm) δ 11.9, 29.8, 121.0, 124.5, 126.1, 128.2, 129.4, 130.5, 132.3, 134.7, 137.6, 146.2, 150.0; MS, m/z (%): 483 [M+ + 2], 305 [M+ − 176], 259 [M+ − 222].N), 1546, 1497 (CC), 1457, 1402, 1369, 1315, 1217, 1135, 1037, 906, 829, 784, 755, 734, 689, 583, 547, 510; 1H NMR (400 MHz, DMSO-d6, ppm) δ 2.31 (s, 6H, 2CH3), 5.56 (s, 1H, CH), 7.20 (m, 2H, Ph), 7.39–7.53 (m, 7H, Ph), 7.78–7.80 (m, 5H, Ph), 7.86–8.02 (m, 3H, Ph).N), 1498 (CC), 1457, 1414, 1368, 1312, 1180, 1074, 1025, 833, 755, 691, 592, 502; 1H NMR (300 MHz, DMSO-d6, ppm) δ 2.28 (s, 6H, 2CH3), 5.10 (s, 1H, CH), 7.24–7.74 (m, 13H), 8.49 (d, J = 7.8 Hz, 1H, Py); 13C NMR (75 MHz, DMSO-d6, ppm) δ 12.3, 36.9, 120.7, 122.1, 122.4, 125.8, 129.3, 137.6, 138.0, 147.1, 148.6, 161.6.N), 1578, 1498 (CC), 1456, 1420, 1355, 1290, 1180, 1102, 1028, 863, 800, 753, 692, 637, 592, 506; 1H NMR (300 MHz, DMSO-d6, ppm) δ 2.34 (s, 6H, 2CH3), 5.04 (s, 1H, CH), 7.24 (t, J = 7.5 Hz, 2H), 7.35 (dd, J = 7.8 Hz, J = 4.8 Hz, 1H), 7.44 (t, J = 7.5 Hz, 4H), 7.70–7.75 (m, 5H), 8.41 (d, J = 4.5 Hz, 1H, Py), 8.50 (d, J = 2.1 Hz, 1H, Py); 13C NMR (75 MHz, DMSO-d6, ppm) δ 12.1, 31.5, 104.2, 120.9, 123.7, 125.9, 129.0, 129.3, 135.5, 137.9, 138.5, 146.6, 147.3, 149.0, 157.5; MS, m/z (%): 260 [M+ − 177], 232 [M+ − 205], 184 [M+ − 253].N), 1499 (CC), 1455, 1411, 1369, 1305, 1243, 1115, 1027, 906, 832, 785, 754, 691, 590, 498; 1H NMR (300 MHz, DMSO-d6, ppm) δ 1.92 (d, J = 13.5 Hz, 3H, CH3), 2.08 (s, 3H, CH3), 2.83 (s, 1H, CH), 7.14–7.50 (m, 10H, Ph), 7.68–7.78 (m, 7H, Ph), 13.85 (br, 1H, OH); 13C NMR (75 MHz, DMSO-d6, ppm) δ 11.2, 14.4, 32.1, 121.1, 126.1, 127.3, 128.5, 129.4, 129.9, 130.7, 132.3, 137.6, 139.8, 146.5; anal. calcd for C29H26N4O2: C, 75.30; H, 5.67; N, 12.11, found: C, 75.26; H, 5.66; N, 12.08%; MS, m/z (%): 285 [M+ − 177], 257 [M+ − 205], 184 [M+ − 278].N), 1525, 1492 (CC), 1380, 1277, 1216, 1134, 1085, 1050, 995, 938, 877, 826, 774, 718, 692, 609, 570, 522, 460, 418; 1H NMR (300 MHz, DMSO-d6, ppm) δ 2.07 (s, 6H, 2CH3), 4.83 (s, 1H, CH), 7.12–7.14 (m, 3H, Ph), 7.19–7.24 (m, 2H, Ph), 11.32 (br, 1H, OH); MS, m/z (%): 283 [M+], 184 [M+ − 100], 127 [M+ − 157].N), 1568, 1533, 1490 (CC), 1470, 1400, 1208, 1167, 1093, 1020, 852, 802, 766, 632, 606, 540, 505, 433, 406; 1H NMR (300 MHz, DMSO-d6, ppm) δ 2.08 (s, 6H, 2CH3), 4.84 (s, 1H, CH), 7.13 (d, J = 8.4 Hz, 2H, Ph), 7.28 (d, J = 8.4 Hz, 2H, Ph), 11.32 (br, 1H, OH); MS, m/z (%): 316 [M+ − 2], 218 [M+ − 100], 184 [M+ − 134].N), 1510, 1456 (CC), 1379, 1286, 1208, 1122, 1085, 1048, 987, 930, 834, 791, 749, 697, 612, 521, 472, 419; 1H NMR (300 MHz, DMSO-d6, ppm) δ 2.08 (s, 6H, 2CH3), 2.24 (s, 3H, PhCH3), 4.80 (s, 1H, CH), 7.05 (s, 4H, Ph); 13C NMR (75 MHz, DMSO-d6, ppm) δ 10.8, 19.0, 20.9, 32.7, 104.8, 127.8, 128.7, 134.6, 140.6, 161.5; anal. calcd for C16H18N4O2: C, 64.41; H, 6.8; N, 18.78, found: C, 64.02; H, 6.61; N, 18.52%; MS, m/z (%): 297 [M+], 199 [M+ − 99], 184 [M+ − 114].O), 1621, 1552, 1521, 1494, 1441, 1374, 1318, 1246, 1189, 1120, 995, 939, 812, 763, 669, 583, 538, 514; 1H NMR (300 MHz, DMSO-d6, ppm) δ 2.29 (s, 3H, CH3), 3.12 (s, 6H, 2NCH3), 6.84 (d, J = 7.5 Hz, 2H), 7.16 (m, 1H), 7.42 (m, 2H), 7.56 (s, 1H), 7.97 (d, J = 7.2 Hz, 2H), 8.65 (d, J = 6.9 Hz, 2H); anal. calcd for C19H19N3O: C, 74.73; H, 6.27; N, 13.76, found: C, 74.00; H, 6.12; N, 13.56%; MS, m/z (%): 305 [M+], 303 [M+ − 2], 171 [M+ − 128].Conclusion
In conclusion, we successfully synthesized HAP@AEPH2-SO3H as a new, green heterogeneous and reusable solid acid catalyst and characterized it using FT-IR, XRD, SEM, TEM and TGA/DTA techniques. It was found that HAP@AEPH2-SO3H particles were rod-like in shape with a mean size range of 10–100 nm. Moreover, characterization results depicted that the nanoHAP was well modified with AEPH2 and sulfonic acid. The new heterogeneous catalyst was used as an efficient solid acid catalyst for rapid synthesis of 4,4′-(aryl methylene)bis(1H-pyrazol-5-ol)s derivatives via the one-pot reaction of phenylhydrazine/hydrazine hydrate, ethyl acetoacetate and aldehydes. The promising points of the present protocol which make this methodology a valid contribution to the existing processes in the field of 4,4′-(aryl methylene)bis(1H-pyrazol-5-ol)s derivative synthesis are efficiency, generality, excellent yields of products, short reaction time, low cost, simple experimental and isolation procedures, cleaner reaction profile and finally compliance with green chemistry protocols.Acknowledgements
The authors gratefully acknowledge the partial support of this study by Ferdowsi University of Mashhad Research Council (Grant no. p/3/29760).References
- B. N. Acharya, D. Saraswat, M. Tiwari, A. K. Shrivastava, R. Ghorpade, S. Apna and M. P. Kaushik, Eur. J. Med. Chem., 2010, 45, 430–438 CrossRef CAS PubMed
.
- M. Kidwai and R. J. Mohan, J. Korean Chem. Soc., 2004, 48, 177–181 CrossRef CAS
- S. Sugiura, S. Ohno, O. Ohtani, K. Izumi, T. Kitamikado, H. Asai, K. Kato, M. Hori and H. Fujimura, J. Med. Chem., 1977, 20, 80–84 CrossRef CAS
- H. Bayrak, A. Demirbas, N. Demirbas and S. A. Karaoglu, Eur. J. Med. Chem., 2010, 45, 4726–4732 CrossRef CAS PubMed
- K. H. Carlsson and I. Jurna, Naunyn-Schmiedeberg’s Arch. Pharmacol., 1987, 335, 154–159 CrossRef CAS
- K. Sujatha, G. Shanthi, N. P. Selvam, S. Manoharan, P. T. Perumal and M. Rajendran, Bioorg. Med. Chem. Lett., 2009, 19, 4501–4503 CrossRef CAS PubMed
- R. V. Antre, A. Cendilkumar, R. Nagarajan, D. Goli and R. J. Oswal, J. Sci. Res., 2012, 4, 183–192 CAS
- M. Londershausen, Pestic. Sci., 1996, 48, 269–292 CrossRef CAS
- R. K. Tewari, R. K. Mishra, S. K. Srivastava and S. C. Bahel, Pestic. Res. J., 1990, 2, 24 Search PubMed
- G. Mariappan, P. B. Saha, L. Sutharson and A. Haldar, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2010, 49, 1671 Search PubMed
- W. S. Hamama, Synth. Commun., 2001, 31, 1335–1345 CrossRef CAS PubMed
- T. Ren, S. Liu, G. Li, J. Zhang, J. Guo, W. Li and L. Yang, Spectrochim. Acta, Part A, 2012, 97, 167–175 CrossRef CAS PubMed
- M. Abbasi-Tarighat, E. Shahbazi and K. Niknam, Food Chem., 2013, 138, 991–997 CrossRef CAS PubMed
- B. S. Kuarm and B. Rajitha, Synth. Commun., 2012, 42, 2382–2387 CrossRef CAS PubMed
- K. R. Phatangare, V. S. Padalkar, V. D. Gupta, V. S. Patil, P. G. Umape and N. Sekar, Synth. Commun., 2012, 42, 1349–1358 CrossRef CAS PubMed
- K. Niknam and S. Mirzaee, Synth. Commun., 2011, 41, 2403–2413 CrossRef CAS PubMed
- S. Sobhani, A. Hasaninejad, M. F. Maleki and Z. P. Parizi, Synth. Commun., 2012, 42, 2245–2255 CrossRef CAS PubMed
- W. Wang, S. X. Wang, X. Y. Qin and J. T. Li, Synth. Commun., 2005, 35, 1263–1269 CrossRef CAS PubMed
- S. Sobhani, E. Safaei, A. Hasaninejad and S. J. Rezazadeh, Organomet. Chem., 2009, 694, 3027–3031 CrossRef CAS PubMed
- A. Hasaninejad, M. Shekouhy, A. Zare, S. M. S. Hoseini Ghattali and N. J. Golzar, J. Iran. Chem. Soc., 2011, 8, 411–423 CrossRef CAS
- E. Mosaddegh, A. Hassankhani and A. J. Baghizadeh, J. Chil. Chem. Soc., 2010, 55, 419–420 CrossRef CAS
- M. A. Gouda and A. A. Abu-Hashem, Green Chem. Lett. Rev., 2012, 5, 203–209 CrossRef CAS PubMed
- Z. Karimi-Jaberi, B. Pooladian, M. Moradi and E. Ghasemi, Chin. J. Catal., 2012, 33, 1945–1949 CrossRef CAS
- S. Tayebi, M. Baghernejad, D. Saberi and K. Niknam, Chin. J. Catal., 2011, 32, 1477–1483 CrossRef CAS
- Y. Hu, P. Wei, H. Zhou, P. K. OuYang and Z. C. Chen, Chin. Chem. Lett., 2006, 17, 299–301 CAS
- S. Sobhani, R. Nasseri and M. Honarmand, Can. J. Chem., 2012, 90, 798–804 CrossRef CAS
- M. Baghernejad and K. Niknam, Int. J. Chem., 2012, 4, 52–60 CAS
- D. Shi, J. Chen, N. Wu, Q. Zhuang and X. Wang, Chin. J. Org. Chem., 2005, 25, 405–408 CAS
- K. Niknam, D. Saberi, M. Sadegheyan and A. Deris, Tetrahedron Lett., 2010, 51, 692–694 CrossRef CAS PubMed
- A. Khazaei, M. A. Zolfigol, A. R. Moosavi-Zare, Z. Asgari, M. Shekouhy, A. Zare and A. Hasaninejad, RSC Adv., 2012, 2, 8010–8013 RSC
- D. Singh and D. Singh, J. Chem. Eng. Data, 1984, 29, 355–356 CrossRef CAS
- A. R. Moosavi-Zare, M. A. Zolfigol, M. Zarei, A. Zare, V. Khakyzadeh and A. Hasaninejad, Appl. Catal., A, 2013, 467, 61–68 CrossRef CAS PubMed
-
(a) A. Hasaninejad, A. Zare, M. Shekouhy and N. Golzar, Org. Prep. Proced. Int., 2011, 43, 131–137 CrossRef CAS PubMed
- M. N. Elinson, A. S. Dorofeev, R. F. Nasybullin and G. I. Nikishin, Synthesis, 2008, 1933–1937 CrossRef CAS
- A. Domling and I. Ugi, Angew. Chem., Int. Ed., 2000, 39, 3168–3210 CrossRef CAS
- C. O. Kappe, Eur. J. Med. Chem., 2000, 35, 1043–1052 CrossRef CAS
-
(a) J. Zhu and H. Bienayme, Multicomponent Reactions, Wiley-VCH, Weinheim, 2005 Search PubMed
- S. Tayebi and K. Niknam, Iran. J. Catal., 2012, 2, 69–74 CAS
- E. Soleimani, S. Ghorbani, M. Taran and A. Sarvary, C. R. Chim., 2012, 15, 955–961 CrossRef CAS PubMed
- A. Khazaei, F. Abbasi and A. R. Moosavi-Zare, New J. Chem., 2014, 38, 5287–5292 RSC
- A. Hasaninejad, M. Rasekhi Kazerooni and A. Zare, ACS Sustainable Chem. Eng., 2013, 1, 679–684 CrossRef
- M. Seddighi, F. Shirini and M. Mamaghani, RSC Adv., 2013, 3, 24046–24053 RSC
- Z. Zhou and Y. Zhang, Green Chem. Lett. Rev., 2014, 7, 18–23 CrossRef PubMed
-
(a) D. Tejedor and F. Garcia-Tellado, Chem. Soc. Rev., 2007, 36, 484–491 RSC
- G. Lelais and D. W. C. MacMillan, Aldrichimica Acta, 2006, 39, 79–87 CAS
- M. Dabiri, P. Salehi, M. Bahramnejad and F. Sherafat, J. Comb. Chem., 2010, 12, 638–642 CrossRef CAS PubMed
-
(a) A. Ethirajan, U. Ziener and K. Landfester, Chem. Mater., 2009, 21, 2218–2225 CrossRef CAS
-
(a) T. Ishikawa, H. Tanaka, A. Yasukawa and K. Kandori, J. Mater. Chem., 1995, 5, 1963–1967 RSC
- A. T. Myller, J. J. Karhe and T. T. Pakkanen, Appl. Surf. Sci., 2010, 257, 1616–1622 CrossRef CAS PubMed
-
(a) N. Razavi and B. Akhlaghinia, RSC Adv., 2015, 5, 12372–12381 RSC
- M. G. Ma, Y. J. Zhu and J. Chang, J. Phys. Chem. B, 2006, 110, 14226–14230 CrossRef CAS PubMed
- Y. Daniels, N. Lyczko, A. Nzihou and S. D. Alexandratos, Ind. Eng. Chem. Res., 2015, 54, 585–596 CrossRef CAS PubMed
- A. Badiei, H. Goldooz, G. Mohammadi Ziarani and A. Abbasi, J. Colloid Interface Sci., 2011, 357, 63–69 CrossRef CAS PubMed
- M. Sheykhan, L. Ma’mani, A. Ebrahimi and A. Heydari, J. Mol. Catal. A: Chem., 2011, 335, 253–261 CrossRef CAS PubMed
- A. Z. Alshemary, Y. Goh, M. Akram, I. R. Razali, M. A. Kadir and R. Hussain, Mater. Res. Bull., 2013, 48, 2106–2110 CrossRef CAS PubMed
-
(a) F. Mohandes and M. Salavati-Niasari, Mater. Sci. Eng., C, 2014, 40, 288–298 CrossRef CAS PubMed
- G. Wang, Y. Zhao, J. Tan, S. Zhu and K. Zhou, Trans. Nonferrous Met. Soc. China, 2015, 25, 490–496 CrossRef CAS
- Y. Jiang, F. Sun, X. Y. Zhou, W. B. Kong and X. Y. Xie, Chin. Chem. Lett., 2015, 26, 1121–1128 CrossRef CAS PubMed
- N. Ahmed and Z. N. Siddiqui, J. Mol. Catal. A: Chem., 2014, 394, 232–243 CrossRef CAS PubMed
- A. D. Gupta, R. Pal and A. K. Mallik, Green Chem. Lett. Rev., 2014, 7, 404–411 CrossRef PubMed
- D. H. Jani, H. S. Patel, H. Keharia and C. K. Modi, Appl. Organomet. Chem., 2010, 24, 99–111 CAS
- B. B. Thummar, U. P. Tarpada and D. K. Raval, J. Heterocycl. Chem., 2014, 51, 1740–1746 CrossRef CAS PubMed
- F. Shirini, M. Seddighi, M. Mazloumi, M. Makhsous and M. Abedini, J. Mol. Liq., 2015, 208, 291–297 CrossRef CAS PubMed
- M. Barge and R. Salunkhe, RSC Adv., 2014, 4, 31177–31183 RSC
- A. Zara, M. Merajoddin, A. R. Moosavi Zare and M. Zarei, Chin. J. Chem., 2014, 35, 85–89 Search PubMed
- J. Safaei Ghomi, B. Khojastehbakht Koopaei and H. Shahbazi Alavi, RSC Adv., 2014, 4, 46106–46113 RSC
- A. Vafaee, A. Davoodnia and M. Pordel, Res. Chem. Intermed., 2015 DOI:10.1007/s11164-014-1896-y
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra16236j |
| This journal is © The Royal Society of Chemistry 2015 |