
Design, synthesis and antifungal activity of carboxylic acid amide fungicides: part 2: substituted 1-phenyl-2-phenoxyethylamino valinamide carbamates†‡
Zhi-Peng Wanga,
Jun-Jie Koub,
Yang Gaoa,
Xiu-Jiang Dua,
Yong-Hong Lia and
Wei-Guang Zhao*a
aState Key Laboratory of Elemento-Organic Chemistry, Collaborative Innovation Center of Chemical Science and Engineering, Nankai University, Tianjin 300071, China. E-mail: zwg@nankai.edu.cn
bNational Pesticide Engineering Research Center (Tianjin), Nankai University, Tianjin 300071, China
First published on 28th August 2015
Abstract
Valinamide carbamates are important agricultural fungicides with anti-oomycete activity and low toxicity toward mammalian cells. To find valinamide carbamates with high activity against resistant pathogens, a series of substituted 1-phenyl-2-phenoxyethylamino derivatives were designed and synthesized by introducing substituted phenyloxy or benzyloxy into valinamide carbamate leads. Bioassays showed that many title compounds exhibited good anti-oomycete activity. Compound 8b had much higher in vitro fungicidal activity against Phytophthora capsici than the control compound iprovalicarb and had a similar potency to the control mandipropamid. At 6.25 μg mL−1 the fungicidal activities of many compounds against in vivo Pseudoperonospora cubensis were greater than 30%, while the control dimethomorph gave only 27% activity at this concentration. The effect of 8b against Pseudoperonospora cubensis in the field indicates promise for a new candidate for controlling cucumber downy mildew.
Introduction
Oomycetes are biologically and biochemically distinct from ascomycetes,1 and exposure to the former can lead to the occurrence of several destructive diseases in a range of important crop plants, such as late blight on potatoes, blue mold on tobacco, and grape downy mildew. Carboxylic acid amide (CAA) fungicides were officially announced by the Fungicide Resistance Action Committee in 2005.1 CAA fungicides exhibit high antifungal activity and a common cross resistance pattern against most foliar oomycete plant pathogens. CAA fungicides can be divided into three different sub-classes based on differences in their structure, including: (1) cinnamic acid amides, such as dimethomorph,2 flumorph,3 and pyrimorph;4 (2) valinamide carbamates, such as benthiavalicarb,5 iprovalicarb,6 and valiphenal;7 and (3) mandelic acid amides, such as mandipropamid.8 A series of studies has shown that the CAA compounds exert fungicidal activity by targeting cellulose synthases in plant pathogen oomycetes.9–14We have previously reported the synthesis of, and evaluated the fungicidal activities of a series of mandelic acid amide and valinamide carbamate derivatives.15–18 We found that all three sub-classes featured similar structural fragments, including amides, halobenzenes (or methylbenzenes) and/or dialkoxyl benzene moieties. Therefore, we designed and synthesized novel valinamide carbamate with all three fragments.19 Compound 1a displayed higher in vitro fungicidal activities against Phytophthora capsici (EC50 0.15 μg mL−1) than iprovalicarb (EC50 0.27 μg mL−1). However, compounds containing a large and sterically bulky halogen or alkyl group showed much lower levels of fungicidal activity. Steric congestion around the N-benzhydryl group may lead to a significant decrease in fungicidal activity.
And it was also reported that two atom spacer in the field of mandelamides, which have been called “stretched mandelamides”, gave excellent fungicidal results.20 Therefore, we synthesized and evaluated the fungicidal activities of a series of “stretched” compounds 2, which bear an OCH2 or CH2OCH2 linker between the 4-methylphenyl ring and the 3,4-dimethoxylphenyl ring of compounds 1, and report the promising antioomycetic activity of the compounds (Fig. 1).
 | ||
| Fig. 1 Design of the target compounds based on compounds 1. | ||
Results and discussion
Chemistry
Compounds 8a–v were synthesized as shown in Scheme 1. The (aryloxy)acetophenones 4a–h were prepared from 2-bromoacetophenone with the corresponding phenol in dry acetone and anhydrous K2CO3. This method was not applied to the synthesis of compounds 4i–o because of a self-substitution reaction of compound 3b. After optimization, it was found that treatment of 2-bromoacetophenone 3b with excess phenol sodium salt (3 equiv.) in DMF at 85–90 °C gave the desired ketones 4i–o in 80–88% yield. The oximes 5a–o were then prepared by addition of NH2OH·HCl and NaOH (R1 = Me) at room temperature or AcONa (R1 = H) at reflux temperature. Using commercial 10% Pd/C, the hydrogenation of the oximes 5a–o readily proceeded to give the corresponding amines (rac)-6a–o. To avoid the loss of chlorine from the phenyl ring, the hydrogenation time of compound 5d was limited to less than 6 hours. Isopropyloxycarbonyl-L-valine was treated with isopropyl chloroformate under basic conditions in tetrahydrofuran to give the mixed anhydride (S)-7, which was not isolated. Treatment with triethylamine and amines (rac)-6a–o in tetrahydrofuran, gave the diastereomeric mixture (R,S)- and (S,S)-8a–o in 80–86% yield. The propargylated title compounds (R,S)- and (S,S)-8p–v (R1 = propargyl) were easily obtained from compounds (rac)-6a–o by alkylation with propargyl bromide.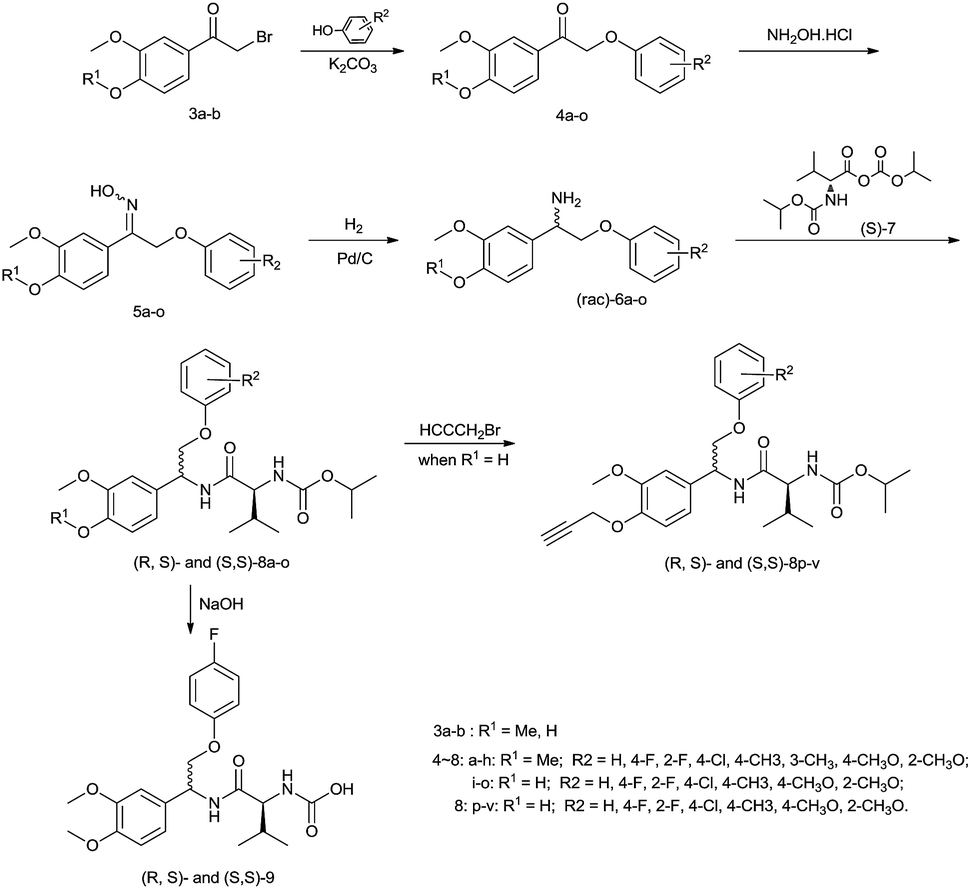 | ||
| Scheme 1 Synthetic route to compounds 8a–v and 9. | ||
Compound (R,S)- and (S,S)-13 was synthesized using a similar method from α-hydroxy ketone 10, which was synthesized by reaction of compound 3a with sodium formate in ethanol, was used instead of the α-phenoxyl ketone 4 (Scheme 2).
 | ||
| Scheme 2 The synthetic route to compounds 14a–f. | ||
The benzylated title compounds (R,S)- and (S,S)-14a–f were easily obtained from compound 12 by alkylation with the corresponding benzyl bromide at −10 °C.
At higher reaction temperatures, treatment of 13 with NaH failed to provide compounds 14a–f, and led to intramolecular N-terminus isopropoxycarbonyl group cleavage by the adjacent amide, preventing alkylation of the hydroxyl group (Scheme 3). Compound 16 was then formed through a benzylation reaction of the intermediate 15.
 | ||
| Scheme 3 Proposed reaction route. | ||
Fungicidal activity
The in vitro fungicidal activities towards Phytophthora capsici of the racemic compounds 8a–h and 14a–f are shown in Table 1 (reported as IC50 values in μg mL−1). The IC50 values were obtained from Petri dish trials and represent the concentrations at which the tested compounds showed 50% inhibitory activity. Interestingly, the introduction of an additional OCH2 or CH2OCH2 linker led to highly active valinamide carbamates 8a–h and 14a–f. These compounds showed impressive efficacy especially against Phytophthora capsici down to 0.043 mg L−1, which was unrivalled by the other valinamide carbamate derivatives.| No. | Substituents | y = a + bx | r2 | EC50 | |||
|---|---|---|---|---|---|---|---|
| R1 | R2 | 0 | μg mL−1 | 95% CIa | |||
| a CI: confidence interval. | |||||||
| 8a | Me | H | 0 | y = 4.4709 + 1.7456x | 0.9526 | 0.23 | 0.20–0.27 |
| 8b | Me | 4-F | 0 | y = 6.0798 + 0.7734x | 0.9887 | 0.043 | 0.038–0.050 |
| 8c | Me | 2-F | 0 | y = 4.3519 + 2.2231x | 0.9865 | 1.96 | 1.60–2.40 |
| 8d | Me | 4-Cl | 0 | y = 6.8320 + 1.7238x | 0.9863 | 0.086 | 0.072–1.104 |
| 8e | Me | 4-Me | 0 | y = 5.7767 + 1.3766x | 0.9609 | 0.27 | 0.22–0.34 |
| 8f | Me | 3-Me | 0 | y = 4.3335 + 2.2087x | 0.9776 | 2.00 | 1.57–2.56 |
| 8g | Me | 4-MeO | 0 | y = 2.6433x + 5.8024 | 0.9825 | 0.49 | 0.28–0.72 |
| 8h | Me | 2-MeO | 0 | y = 5.3894 + 1.5397x | 0.9547 | 0.55 | 0.36–0.84 |
| 8i | H | H | 0 | y = 4.2685 + 2.5376x | 0.9624 | 1.94 | 1.41–2.68 |
| 8j | H | 4-F | 0 | y = 6.9321 + 3.8425x | 0.9700 | 0.35 | 0.25–0.49 |
| 8j-Na | Na | 4-F | 0 | y = 5.5929 + 1.5648x | 1.0599 | 0.78 | 0.61–1.06 |
| 8k | H | 2-F | 0 | y = 4.2220 + 2.2140x | 0.9717 | 2.25 | 1.69–2.99 |
| 8l | H | 4-Cl | 0 | y = 6.0769 + 2.2965x | 0.9857 | 0.34 | 0.28–0.40 |
| 8m | H | 4-Me | 0 | y = 4.6161 + 2.3713x | 0.9864 | 1.45 | 1.21–1.75 |
| 8n | H | 4-MeO | 0 | y = 4.7523 + 2.0574x | 0.9754 | 1.32 | 1.02–1.70 |
| 8o | H | 2-MeO | 0 | y = 3.8688 + 2.5226x | 0.9539 | 2.81 | 1.98–3.99 |
| 8p | Propargyl | H | 0 | y = 4.0813 + 1.3259x | 0.9836 | 4.93 | 3.90–6.23 |
| 8q | Propargyl | 4-F | 0 | y = 6.0914 + 1.8758x | 0.9594 | 0.26 | 0.19–0.37 |
| 8r | Propargyl | 2-F | 0 | y = 4.9922 + 2.9204x | 0.9814 | 1.04 | 0.82–1.33 |
| 8s | Propargyl | 4-Cl | 0 | y = 4.3558 + 0.6022x | 0.9555 | 11.74 | 6.99–19.73 |
| 8t | Propargyl | 4-Me | 0 | y = 4.1512 + 0.8110x | 0.9964 | 11.13 | 9.68–12.80 |
| 8u | Propargyl | 4-MeO | 0 | y = 5.076 + 2.5299x | 0.9598 | 0.93 | 0.66–1.31 |
| 8v | Propargyl | 2-MeO | 0 | y = 4.3886 + 0.6946x | 0.9953 | 7.59 | 6.60–8.73 |
| 9 | Me | 4-F | 0 | y = 3.4574 + 1.1895x | 0.9663 | 39.63 | 25.07–62.65 |
| 14aa | Me | H | 1 | y = 6.7994 + 3.2111x | 0.9417 | 0.33 | 0.26–0.41 |
| 14ba | Me | 4-F | 1 | y = 4.4663 + 1.5062x | 0.9203 | 2.26 | 1.36–3.74 |
| 14ca | Me | 3-F | 1 | y = 5.5754 + 1.9131x | 0.9996 | 0.50 | 0.48–0.53 |
| 14da | Me | 2-F | 1 | y = 5.3742 + 1.4938x | 0.9960 | 0.56 | 0.35–0.90 |
| 14ea | Me | 4-Cl | 1 | y = 3.5790 + 1.6754x | 0.9880 | 7.05 | 5.86–8.48 |
| 14fa | Me | 4-Me | 1 | y = 5.3578 + 1.9975x | 0.9830 | 0.66 | 0.50–0.87 |
| Dimethomorph | y = 9.1342 + 6.3811x | 0.9930 | 0.12 (0.10–0.24) | ||||
| Iprovalicarb | y = 6.2088 + 2.1245x | 0.9966 | 0.27 (0.22–0.33) | ||||
| Mandipropamid | y = 8.4774 + 2.2311x | 0.9685 | 0.04 (0.03–0.06) | ||||
Most of the synthesized compounds 8a–h (R1 = Me, n = 0) showed excellent fungicidal activity against Phytophthora capsici and structure–activity relationship analysis indicated that the substituents R2 had a significant impact on the activity. Compounds containing small groups at the para-position of the phenyl ring (e.g., 8b, R2 = 4-F; 8e, R2 = 4-Me; 8g, R2 = 4-MeO), gave higher levels of fungicidal activity against Phytophthora capsici than those substituted at the meta- (e.g., 8c, R2 = 3-F; 8f, R2 = 3-Me) and ortho-positions (e.g., 8h, R2 = 2-MeO). Compounds with halogens showed higher levels of antioomycetic activity than compounds with alkyl or alkoxyl groups at the same position. The intrinsic order of fungicidal activity of the substituents (R1) was F > Cl ≫ Me > H ≈ MeO. Furthermore, compound 8b (IC50 = 0.043 μg mL−1) and 8d (IC50 = 0.086 μg mL−1) showed higher antioomycetic potency than iprovalicarb (IC50 = 0.27 μg mL−1) and dimethomorph (IC50 = 0.23 μg mL−1), and 8b showed a level of potency very similar to that of mandipropamid (IC50 = 0.024 μg mL−1).
A further in vivo assay was conducted in a greenhouse to estimate the fungicidal activities of the most active compounds against Phytophthora capsici and Pseudoperonospora cubensis. The title compounds were applied to pepper plants through a root irrigation mode, because Phytophthora capsici is a soil-borne disease. As shown in Table 2, the compounds showed lower levels of in vivo fungicidal activity against Phytophthora capsici than the control compounds dimethomorph and iprovalicarb. Under equivalent dosage conditions of 50 mg L−1, compounds 8d, 8e and 8h showed 100% inhibition against Phytophthora capsici, whereas compound 8b, which was identified as the most potent compound from the in vitro fungicidal assay, showed a slightly lower level of inhibition (96%). The fungicidal activities of all of the test compounds were lower against Phytophthora capsici at concentrations of 12.5 μg mL−1, whereas the control compounds still showed 100% inhibition at this concentration. High c![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logP values represent a major stumbling block for drug absorption through the root, so we calculated the clogP values (octanol/water) of the test compounds using SYBYL. The results shown in Table 2 indicate that the introduction of a second phenyl ring into the lead compounds led to a significant increase in their clogP values. All of the compounds 8a–h (clogP > 4.22) had much higher clogP values than iprovalicarb (clogP = 3.2) and dimethomorph (clogP = 2.73). However, compounds with a high clogP values would tight bind to the waxy layer of the leaf and provide a highly effective, weatherproof and long-lasting barrier to disease if applied by foliar spray. Most of the compounds 8a–h in Table 2 exhibited very good in vivo inhibition against Pseudoperonospora cubensis. At 6.25 μg mL−1 compounds 8d and 8e displayed significant levels of control against Pseudoperonospora cubensis, of 47% and 44%, respectively, and showed higher levels of antioomycetic activity than dimethomorph (27%) at the same concentration.
logP values represent a major stumbling block for drug absorption through the root, so we calculated the clogP values (octanol/water) of the test compounds using SYBYL. The results shown in Table 2 indicate that the introduction of a second phenyl ring into the lead compounds led to a significant increase in their clogP values. All of the compounds 8a–h (clogP > 4.22) had much higher clogP values than iprovalicarb (clogP = 3.2) and dimethomorph (clogP = 2.73). However, compounds with a high clogP values would tight bind to the waxy layer of the leaf and provide a highly effective, weatherproof and long-lasting barrier to disease if applied by foliar spray. Most of the compounds 8a–h in Table 2 exhibited very good in vivo inhibition against Pseudoperonospora cubensis. At 6.25 μg mL−1 compounds 8d and 8e displayed significant levels of control against Pseudoperonospora cubensis, of 47% and 44%, respectively, and showed higher levels of antioomycetic activity than dimethomorph (27%) at the same concentration.
| No. | P. capsici (% control at given concentration mg L−1) | P. cubensis (% control at given concentration mg L−1) | clogP |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 100 | 50 | 25 | 12.5 | 6.25 | 100 | 50 | 25 | 12.5 | 6.25 | ||
| a No test. | |||||||||||
| 8a | 100 | 96 ± 4 | 21 ± 5 | 21 ± 4 | 0 | 72 ± 2 | 68 ± 3 | 62 ± 3 | 52 ± 2 | 37 ± 4 | 4.49 |
| 8b | 100 | 96 ± 4 | 93 ± 3 | 25 ± 5 | 0 | 84 ± 1 | 78 ± 2 | 61 ± 2 | 54 ± 3 | 47 ± 4 | 4.77 |
| 8c | 100 | 45 ± 6 | 42 ± 5 | 25 ± 3 | 21 ± 5 | 58 ± 3 | 40 ± 2 | —a | — | — | 4.57 |
| 8d | 100 | 100 | 100 | 32 ± 4 | 23 ± 4 | 84 ± 3 | 79 ± 2 | 78 ± 3 | 53 ± 5 | 34 ± 5 | 5.34 |
| 8e | 100 | 100 | 87 ± 3 | 30 ± 5 | 18 ± 6 | 82 ± 1 | 80 ± 3 | 72 ± 2 | 62 ± 3 | 44 ± 3 | 4.98 |
| 8f | 92 ± 2 | 62 ± 3 | 54 ± 3 | 40 ± 5 | 23 ± 5 | 42 ± 3 | 25 ± 5 | — | — | — | 4.98 |
| 8g | 92 ± 3 | 63 ± 2 | 42 ± 5 | 25 ± 3 | 21 ± 4 | 84 ± 3 | 75 ± 2 | 50 ± 2 | 44 ± 4 | 30 ± 3 | 4.57 |
| 8h | 100 | 100 | 0 | 0 | 0 | 70 ± 4 | 60 ± 2 | 52 ± 2 | 35 ± 3 | 29 ± 2 | 4.22 |
| 8i | 71 ± 3 | 63 ± 4 | 54 ± 5 | 42 ± 4 | 33 ± 3 | 79 ± 2 | 68 ± 3 | 54 ± 2 | 49 ± 4 | 35 ± 3 | 4.01 |
| 8j | 100 | 71 ± 2 | 65 ± 3 | 63 ± 5 | 46 ± 4 | 76 ± 4 | 70 ± 2 | 65 ± 3 | 47 ± 5 | 17 ± 6 | 4.29 |
| 8j-Na | 100 | 100 | 100 | 90 ± 5 | 75 ± 3 | 78 ± 2 | 72 ± 5 | 60 ± 3 | 48 ± 4 | 34 ± 4 | 3.45 |
| 8k | 96 ± 4 | 75 ± 4 | 46 ± 4 | 42 ± 3 | 29 ± 5 | 79 ± 1 | 69 ± 3 | 58 ± 3 | 44 ± 4 | 38 ± 3 | 4.09 |
| 8l | 66 ± 3 | 49 ± 3 | 49 ± 3 | 45 ± 5 | 36 ± 4 | 81 ± 3 | 70 ± 2 | 63 ± 5 | 58 ± 2 | 36 ± 4 | 4.86 |
| 8m | 79 ± 2 | 75 ± 3 | 49 ± 4 | 32 ± 5 | 23 ± 3 | 82 ± 5 | 72 ± 2 | 63 ± 2 | 48 ± 3 | 37 ± 4 | 4.51 |
| 8n | 96 ± 3 | 71 ± 2 | 50 ± 3 | 46 ± 4 | 29 ± 3 | 83 ± 2 | 72 ± 3 | 63 ± 3 | 50 ± 4 | 30 ± 4 | 4.10 |
| 8o | 96 ± 4 | 88 ± 2 | 58 ± 4 | 55 ± 2 | 25 ± 3 | 80 ± 3 | 68 ± 1 | 58 ± 3 | 40 ± 3 | 35 ± 5 | 3.75 |
| 8p | 63 ± 5 | — | — | — | — | — | — | — | — | — | 5.09 |
| 8q | 100 | 97 ± 3 | 16 ± 3 | 0 | 0 | 75 ± 3 | 62 ± 3 | 54 ± 2 | — | — | 5.37 |
| 8r | 75 ± 2 | 67 ± 2 | 50 ± 5 | 38 ± 4 | 34 ± 5 | 70 ± 5 | 65 ± 2 | 47 ± 1 | — | — | 5.17 |
| 8s | 79 ± 5 | — | — | — | — | — | — | — | — | — | 5.94 |
| 8t | 63 ± 3 | — | — | — | — | — | — | — | — | — | 5.58 |
| 8u | 88 ± 5 | — | — | — | — | 68 ± 2 | 57 ± 3 | 42 ± 5 | — | — | 5.17 |
| 8v | 50 ± 5 | — | — | — | — | — | — | — | — | — | 4.82 |
| 9 | 29 ± 4 | — | — | — | — | 23 ± 6 | — | — | — | — | 2.85 |
| 14a | 100 | 87 ± 4 | 78 ± 4 | 52 ± 3 | 48 ± 5 | 84 ± 3 | 72 ± 1 | 66 ± 3 | 60 ± 3 | 42 ± 4 | 4.43 |
| 14b | 100 | 100 | 70 ± 5 | 61 ± 3 | 48 ± 2 | 79 ± 2 | 69 ± 2 | 58 ± 4 | 53 ± 3 | 43 ± 2 | 4.57 |
| 14c | 100 | 83 ± 2 | 65 ± 3 | 31 ± 5 | 7 ± 7 | 72 ± 3 | 69 ± 2 | 63 ± 1 | 47 ± 4 | 32 ± 5 | 4.57 |
| 14d | 100 | 100 | 83 ± 5 | 48 ± 4 | 31 ± 6 | 76 ± 2 | 67 ± 1 | 63 ± 2 | 42 ± 3 | 27 ± 6 | 4.57 |
| 14e | 100 | 100 | 83 ± 2 | 65 ± 3 | 52 ± 3 | 71 ± 4 | 66 ± 4 | 52 ± 3 | 42 ± 3 | 31 ± 5 | 5.14 |
| 14f | 100 | 96 ± 4 | 74 ± 3 | 65 ± 5 | 48 ± 4 | 73 ± 1 | 64 ± 3 | 57 ± 3 | 54 ± 5 | 42 ± 4 | 4.93 |
| Iprovalicarb | 100 | 100 | 100 | 100 | 71 ± 3 | — | — | — | — | — | 3.2 |
| Dimethomorph | 100 | 100 | 100 | 100 | 85 ± 2 | 82 ± 2 | 74 ± 3 | 54 ± 3 | 37 ± 2 | 27 ± 5 | 2.73 |
To reduce the clogP value of the compounds, phenol derivatives 8i–o (R1 = H) and an acid derivative 9 were synthesized. Regrettably the ester group of carbamates negatively influenced the antioomycetic activity. Both benthiavancarb (acid) and benthiavancarb–isopropyl (ester) exhibited excellent fungicidal activities, compared with the acid derivative 9 (EC50 = 39.63 μg mL−1). The phenol derivatives 8i–o maintained excellent in vitro fungicidal activity against Phytophthora capsici and showed the same structure–activity relationship. Compound 8j (EC50 = 0.35 μg mL−1) and 8l (EC50 = 0.34 μg mL−1) showed very similar potency to iprovalicarb (EC50 = 0.27 μg mL−1). However, losing the methyl group did not significantly decrease their clogP values or increase in vivo fungicidal activities against Phytophthora capsici. Nearly all phenol derivatives 8i–o showed higher levels of in vivo Pseudoperonospora cubensis activity than dimethomorph. To further increase the hydrophilic character of the phenol derivatives, the sodium phenate derivative 8j-Na was prepared to test its fungicidal activities. The sodium phenates had lower in vitro fungicidal activity (8j-Na: EC50 = 0.78 μg mL−1), but significantly increased in vivo fungicidal activity against Phytophthora capsici. 8i-Na (75%) showed a level of potency very similar to that of iprovalicarb (71%) and dimethomorph (85%) at 12.5 μg mL−1.
The introduction of a propargyloxy functionality into mandelic acid amides (such as mandipropamid) leads to an enormous increase in fungicidal efficacy. Propargylation of the free hydroxyl functionality of the phenol derivatives 8i–o was attempted to increase fungicidal activity. Propargyl-substituted compounds 8p–v remained quite active (in vitro), but were clearly weaker than their methyl-substituted counterparts (8a–h). This result may be attributed to an adverse steric effect from the propargyl group in vitro activity. Further increases in lipophilicity decreased in vivo activity against Phytophthora capsici.
The highly stretched compounds 14a–f with a three-atom spacer were still highly active toward Phytophthora capsici (in vitro) and Pseudoperonospora cubensis (in vivo). Both subclasses featured similar structure–activity relationships. Interestingly, para-substituted compounds had a slightly lower activity than counterparts with a two-atom spacer; unsubstituted or 2-fluoro substituted compounds had slightly higher activity than counterparts with a two-atom spacer. Unsubstituted compound 14a showed a level of potency (in vitro) very similar to iprovalicarb and dimethomorph. This result may be also attributed to an adverse steric effect from para substitution. The intrinsic order of fungicidal activity of the substituents (R2) was H > F > Cl. All compounds 14a–f showed higher levels of in vivo Pseudoperonospora cubensis activity than dimethomorph.
Field evaluation
Compound 8b, which exhibited excellent fungicidal activity in the laboratory, was chosen to evaluate its fungicidal activity in the field. Field trials were carried out in 2012 and 2013 in Tianjin city and Shandong province. The results of the field trials are shown in Table 3. The field trial in Shandong showed that 8b at 500 mg kg−1, or even lower concentrations (167 or 100 mg L−1), had the same control effect as dimethomorph at 500 mg L−1. The control efficiencies of 8b at 500 and 167 mg L−1 were 73.1 and 68.7%, respectively. The field trial in Tianjin showed that the control efficiencies of 8b at 167 and 100 mg L−1 were 80.5 and 76.6%, respectively. The control efficiencies of dimethomorph at 500 mg L−1 were 73.5 and 79.4% in Shandong and Tianjin, respectively. Furthermore, it was found that compound 8b had a beneficial protective effect on new leaves in the field. The field trials suggest that 8b could be considered for further development as an alternative to the excessively used dimethomorph.| Trial sites | Treatments | Dosage (mg L−1) | % Effectb |
|---|---|---|---|
| a The amount sprayed is about 1125 L ha−1.b Values followed by the same letter within a column are not significantly different at the P < 0.05 level. | |||
| Shandong provincea (2012) | 8b | 500 | 73.1a |
| 167 | 68.7a | ||
| Dimethomorph | 500 | 73.5a | |
| Tianjin cityb (2013) | 8b | 167 | 80.5b |
| 100 | 76.6a | ||
| Dimethomorph | 500 | 79.4ab | |
Experimental
Material and methods
1H and 13C NMR spectra were measured on a Bruker AC-P500 instrument using TMS as internal standard and CDCl3 as solvent. Melting points were determined on an X-4 binocular microscope melting point apparatus (Beijing Tech Instruments, Beijing, China) and were uncorrected. HRMS were recorded on an Ionspec 7.0 T Fourier-transform ion-cyclotron resonance (FTICR) mass spectrometer. All reagents are analytical grade.5a. White solid, yield 84.1%, mp 85–87 °C. 1H NMR (400 MHz, CDCl3) δ 7.57 (dt, J = 21.4, 10.7 Hz, 1H, Ar-H), 7.50 (d, J = 1.9 Hz, 1H, Ar-H), 7.27–7.17 (m, 1H, Ar-H), 6.94–6.78 (m, 4H, Ar-H), 5.16 (s, 2H, CH2), 3.87 (d, J = 8.9 Hz, 6H, Ar-m-OCH3 + Ar-p-OCH3); 13C NMR (101 MHz, CDCl3) δ 193.20, 158.10, 153.92, 149.28, 129.59, 127.78, 122.86, 121.60, 114.81, 110.36, 110.15, 70.71, 56.15, 56.05.
The data for compounds 5b–h can be found in the ESI.‡
5i. White solid, yield 78.2%, mp 67–68 °C. 1H NMR (400 MHz, CDCl3) δ 7.26 (m, 4H, Ar-H), 7.03–6.77 (m, 4H, Ar-H), 5.41–4.81 (m, 2H, CH2), 3.90 (d, J = 4.8 Hz, 3H, CH3); 13C NMR (101 MHz, CDCl3) δ 158.08, 155.65, 147.19, 146.42, 129.55, 125.52, 121.37, 121.19, 114.64, 114.27, 109.26, 59.69, 55.99.
The data for compounds 5j–o can be found in the ESI.‡
6a. Light yellow solid, yield 78.7%, mp 77–78 °C. 1H NMR (300 MHz, CDCl3) δ 7.29 (dd, J = 9.5, 6.3 Hz, 2H, Ar-H), 7.10–6.78 (m, 6H, Ar-H), 4.41 (dd, J = 8.9, 3.5 Hz, 1H, CHCH2), 4.07 (dd, J = 9.0, 3.6 Hz, 1H, CHCH2), 3.91 (d, J = 7.3 Hz, 7H, Ar-m-OCH3 + Ar-p-OCH3 + NH2CH), 2.26–1.71 (m, 2H, NH2).
The data for compounds 6b–o can be found in the ESI.‡
8a. White solid, yield 80.4%, mp 138–140 °C. 1H NMR (400 MHz, CDCl3) δ 7.37–7.20 (m, 2H, Ar-H), 7.02–6.75 (m, 6H, Ar-H), 6.67 (s, 1H, CHCONH), 5.32 (s, 1H, OCONH), 5.16 (s, 1H, Ar-CH), 4.86 (td, J = 12.1, 5.9 Hz, 1H, OCH(CH3)2), 4.34–4.10 (m, 2H, OCH2), 4.00 (dd, J = 17.6, 11.0 Hz, 1H, OCONHCH), 3.86 (s, 6H, Ar-m-OCH3 + Ar-p-OCH3), 2.17 (d, J = 5.9 Hz, 1H, CHCH(CH3)2), 1.25–1.10 (m, 6H, OCH(CH3)2), 1.05–0.83 (m, 6H, CHCH(CH3)2); 13C NMR (101 MHz, CDCl3) δ 171.15, 158.26, 156.26, 149.14, 148.59, 131.36, 129.58, 121.40, 118.99, 114.59, 111.08, 110.27, 69.90, 68.71, 60.52, 55.89, 52.30, 31.08, 22.00, 19.11, 18.03; HRMS (MALDI) m/z calcd for C25H34N2O6Na+ [M + Na]+ 495.2466, found 495.2463.
The data for compounds 8b–o can be found in the ESI.‡
8p. Light yellow solid, yield 47.2%, mp 155–157 °C. 1H NMR (400 MHz, CDCl3) δ 6.99–6.81 (m, 5H, Ar-H), 6.79–6.66 (m, 2H, Ar-H), 6.56 (d, J = 6.6 Hz, 1H, CHCONH), 5.23 (s, 1H, OCONH), 5.04 (s, 1H, Ar-CH), 4.80 (dt, J = 12.5, 6.3 Hz, 1H, OCH(CH3)2), 4.68 (d, J = 2.3 Hz, 2H, CHCCH2), 4.12 (qd, J = 9.7, 5.0 Hz, 2H, OCH2Ar), 3.92 (dd, J = 17.4, 9.3 Hz, 1H, OCONHCH), 3.79 (s, 3H, C6H3OCH3), 2.43 (t, J = 2.3 Hz, 1H, CH2CCH), 2.10 (d, J = 5.8 Hz, 1H, CHCH(CH3)2), 1.21–1.02 (m, 6H, OCH(CH3)2), 0.96–0.76 (m, 6H, CHCH(CH3)2); 13C NMR (101 MHz, CDCl3) δ 170.13, 157.74, 155.32, 153.34, 148.68, 145.36, 131.77, 117.73, 115.03, 114.80, 113.22, 109.85, 77.37, 74.85, 69.58, 67.67, 59.37, 55.71, 54.75, 50.97, 29.87, 21.09, 18.34; HRMS (MALDI) m/z calcd for C17H16F3NO3Na+ [M + Na]+ 523.2215, found 523.2218.
The data for compounds 8q–v can be found in the ESI.‡
9. Light yellow solid, yield 97.2%, mp 75–78 °C. 1H NMR (400 MHz, CDCl3) δ 6.93 (t, J = 8.5 Hz, 4H, Ar-H), 6.86–6.74 (m, 3H, Ar-H), 6.04 (d, J = 24.4 Hz, 1H, CHCONH), 5.38 (s, 1H, OCONH), 4.95 (d, J = 24.5 Hz, 1H, Ar-CH), 4.35–4.16 (m, 1H, OCONHCH), 4.14–4.01 (m, 2H, CHCH2O), 3.84 (s, 6H, Ar-m-OCH3 + Ar-p-OCH3), 2.18–2.03 (m, 1H, CHCH(CH3)2), 0.81 (ddd, J = 35.2, 28.0, 6.6 Hz, 6H, CHCH(CH3)2); 13C NMR (101 MHz, CDCl3) δ 176.10, 158.58, 156.34, 154.28, 149.23, 148.75, 131.69, 119.10, 116.00, 115.81, 111.36, 110.25, 71.72, 58.42, 55.90, 54.03, 30.61, 19.09, 17.55; HRMS (MALDI) m/z calcd for C27H38N2O6Na+ [M + Na]+ 457.1745, found 457.1748.
(rac)-13. White solid, yield: 81.8%, mp: 179–183 °C. 1H NMR (300 MHz, CDCl3) δ 7.09–6.59 (m, 4H, Ar-H + CHCONH), 5.32 (dd, J = 45.8, 8.1 Hz, 1H, OCONH), 4.96 (d, J = 4.8 Hz, 1H, Ar-CH), 4.77 (d, J = 5.8 Hz, 1H, OCH(CH3)2), 3.78 (s, 8H, CHCH2O + Ar-m-OCH3 + Ar-p-OCH3), 2.14–1.90 (m, 1H, CHCH(CH3)2), 1.15 (d, J = 5.6 Hz, 6H, OCH(CH3)2), 0.99–0.75 (m, 6H, CHCH(CH3)2); 13C NMR (101 MHz, CDCl3) δ 172.01, 156.41, 148.87, 148.18, 131.70, 118.49, 110.93, 110.01, 68.41, 66.03, 63.32, 55.80, 53.72, 30.88, 21.94, 19.44, 17.59; HRMS (MALDI) m/z calcd for C19H30N2O6Na+ [M + Na]+ 405.1996, found 405.1999.
(R,S)- and (S,S)-14a. White solid, yield 54.9%, mp 147–149 °C. 1H NMR (400 MHz, CDCl3) δ 7.32 (dt, J = 13.5, 5.6 Hz, 5H, Ar-H), 6.86 (d, J = 13.9 Hz, 3H, Ar-H), 6.67 (d, J = 7.4 Hz, 1H, CHCONH), 5.21 (d, J = 8.2 Hz, 1H, OCONH), 5.15 (d, J = 2.7 Hz, 1H, Ar-CH), 4.91 (s, 1H, OCH(CH3)2), 4.62–4.46 (m, 2H, C6H4CH2O), 4.01 (dd, J = 13.4, 7.1 Hz, 1H, OCONHCH), 3.91–3.82 (m, 6H, Ar-m-OCH3 + Ar-p-OCH3), 3.74 (d, J = 4.2 Hz, 2H, CHCH2O), 2.14 (s, 1H, CHCH(CH3)2), 1.37–1.11 (m, 6H, OCH(CH3)2), 1.04–0.81 (m, 6H, CHCH(CH3)2); 13C NMR (101 MHz, CDCl3) δ 170.59, 156.03, 148.83, 148.17, 137.36, 132.17, 128.38, 127.85, 127.68, 118.74, 111.10, 110.32, 73.18, 72.06, 68.57, 60.39, 55.90, 52.27, 31.20, 22.08, 19.25, 17.66; HRMS (MALDI) m/z calcd for C26H36N2O6Na+ [M + Na]+ 495.2466, found 495.2458.
The data for compounds 14b–f can be found in the ESI.‡
Fungicidal activities
Conclusions
In previous studies, we found that valinamide carbamate derivatives of 1 with three active structural fragments display promising antioomycetic activity. Herein, we designed and synthesized a series of “stretched” compounds with OCH2 or CH2OCH2 spacers that increased the flexibility between the two benzene rings of the three fragments. Evaluation of the fungicidal activities of these compounds revealed that these “stretched” compounds possessed much higher fungicidal activities against Phytophthora capsici and Pseudoperonospora cubensis than the lead compounds 1. In vitro data showed compounds 8b and 8d were also more potent against the control compounds dimethomorph and iprovalicarb, and 8b had nearly equivalent activity to mandipropamid. In vivo data showed most of the “stretched” compounds had higher antioomycetic potency against Pseudoperonospora cubensis than dimethomorph. The field trial results from two sites in different parts of China clearly showed that 8b had nearly equivalent activity against Pseudoperonospora cubensis at a 100 mg L−1 dose to dimethomorph at a 500 mg L−1 dose. Because of the easy preparation and good antioomycetic activity, and unique mechanism of action, 8b is a promising candidate for future development. Further research on acute toxicity, field residues, and their inhibitory activities against other oomycetes is ongoing and will be reported in the future.Acknowledgements
We are grateful for financial support for this work from the National Natural Science Foundation of China (21172124).Notes and references
- W. Krämer, U. Schirmer, P. Jeschke, M. Witschel, Modern Crop Protection Compounds, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2012 Search PubMed.
- G. Albert, J. Curtze and C. A. Drandarevski, Brighton Crop Prot. Conf.--Pests Dis., 1988, 17–24 CAS.
- C. Liu, W. Liu and Z. Li, The BCPC Conference: Pests and diseases, Proceedings of an international conference held at the Brighton Hilton Metropole Hotel, Brighton, UK, 2000, vol. 2 Search PubMed.
- Z. Qin, C. Mu, S. Mao, Y. Dong, N. Li and S. Zhang, China Patent CN 566095, 2005.
- Y. Miyake, J. Sakai, I. Miura, K. Nagayama and M. Shibata, The BCPC International Congress: Crop Science and Technology, Volumes 1 and 2, Proceedings of an international congress held at the SECC, Glasgow, Scotland, UK, 2003 Search PubMed.
- R. P. K. Stenzel, T. Seitz and A. W. R. Tiemann, Brighton Crop Prot. Conf.--Pests Dis., 1998, 367–374 Search PubMed.
- G. Agosteo, E. Marsilii, A. Pane, C. Rizza, F. Raudino, S. Cacciola, A. Giambelli and G. d. San Lio, Strategie innovative di difesa nel settore ortoflorofrutticolo, Torino, Italia, 2010 Search PubMed.
- C. Lamberth, A. Jeanguenat, F. Cederbaum, A. De Mesmaeker, M. Zeller, H. Kempf and R. Zeun, Bioorg. Med. Chem., 2008, 16, 1531–1545 CrossRef CAS PubMed.
- M. Blum, M. Waldner and U. Gisi, Fungal Genet. Biol., 2010, 47, 499–510 CrossRef CAS PubMed.
- M. Blum, M. Boehler, E. Randall, V. Young, M. Csukai, S. Kraus, F. Moulin, G. Scalliet, A. O. Avrova, S. C. Whisson and R. Fonne-Pfister, Mol. Plant Pathol., 2010, 11, 227–243 CrossRef CAS PubMed.
- M. Blum, M. Waldner, G. Olaya, Y. Cohen, U. Gisi and H. Sierotzki, Pest Manage. Sci., 2011, 67, 1211–1214 CrossRef CAS PubMed.
- Y. Aoki, S. Furuya and S. Suzuki, Pest Manage. Sci., 2011, 67, 1557–1561 CrossRef CAS PubMed.
- Z. Pang, J. Shao, L. Chen, X. Lu, J. Hu, Z. Qin and X. Liu, PLoS One, 2013, 8, e56513 CAS.
- M. Blum, M. Waldner, G. Olaya, Y. Cohen, U. Gisi and H. Sierotzki, Pest Manage. Sci., 2011, 67, 1211–1214 CrossRef CAS PubMed.
- N. Su, Z. J. Wang, L. Z. Wang, X. Zhang, W. L. Dong, H. X. Wang, Z. M. Li and W. G. Zhao, Chem. Biol. Drug Des., 2011, 78, 101–111 CAS.
- H. H. Li, Z. J. Wang, L. Z. Wang, Z. M. Li and W. G. Zhao, Chem. J. Chin. Univ., 2011, 32, 79–83 CAS.
- S. Li, C. Cui, M. Y. Wang, S. J. Yu, Y. X. Shi, X. Zhang, Z. M. Li, W. G. Zhao and B. J. Li, J. Fluorine Chem., 2012, 137, 108–112 CrossRef CAS PubMed.
- H. W. Yao, H. H. Li, N. N. Su, H. X. Wang, X. H. Liu, L. Z. Wang, X. Zhang, Z. M. Li and W. G. Zhao, Chem. J. Chin. Univ., 2009, 30, 908–912 CAS.
- X. J. Du, Q. Bian, H. X. Wang, S. Yu, J. J. Kou, Z. P. Wang, Z. M. Li and W. G. Zhao, Org. Biomol. Chem., 2014, 12, 5427–5434 CAS.
- C. Lamberth, H. J. Kempf and M. Križ, Pest Manage. Sci., 2007, 63, 57–62 CrossRef CAS PubMed.
- L. Hurrell, L. Johnston, N. Mathivanan and D. Vong, Can. J. Chem., 1993, 71, 1340–1348 CrossRef CAS.
- W. H. Pirkle and K. A. Simmons, J. Org. Chem., 1983, 48, 2520–2527 CrossRef CAS.
- N. N. Su, L. X. Xiong, S. J. Yu, X. Zhang, C. Cui, Z. M. Li and W. G. Zhao, Comb. Chem. High Throughput Screening, 2013, 16, 484–493 CrossRef CAS.
- J. Xu, X. Zhao, X. Han and Y. Du, Pestic. Biochem. Physiol., 2007, 87, 220–228 CrossRef CAS PubMed.
- S. Mitani, S. Araki, T. Yamaguchi, Y. Takii, T. Ohshima and N. Matsuo, Pestic. Biochem. Physiol., 2001, 70, 92–99 CrossRef CAS.
- J. Andrés Ares, A. Rivera Martínez and J. Fernández Paz, Span. J. Agric. Res., 2005, 3, 429–436 CrossRef.
- M. Li, C. L. Liu, L. Li, H. Yang, Z. N. Li, H. Zhang and Z. M. Li, Pest Manage. Sci., 2010, 66, 107–112 CrossRef CAS PubMed.
Footnotes |
| † Part 1: design, synthesis, and fungicide activities of novel carboxylic acid amides represented by N-benzhydryl valinamode carbamates, Org. Biomol. Chem., 2014, 12, 5427–5434. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra16225d |
| This journal is © The Royal Society of Chemistry 2015 |