DOI:
10.1039/C5RA16114B
(Paper)
RSC Adv., 2015,
5, 79556-79564
Facile synthesis of CeO2 nanoparticle sensitized CdS nanorod photocatalyst with improved visible-light photocatalytic degradation of rhodamine B†
Received
11th August 2015
, Accepted 4th September 2015
First published on 4th September 2015
Abstract
A heterostructure photocatalyst consisting of one-dimensional (1D) CdS nanorods (NRs) and cerium dioxide (CeO2) nanoparticles (NPs) was successfully synthesized via a solvothermal method. Different characterization techniques confirmed that CeO2 NPs were intimately attached on CdS NRs. The visible-light-driven photocatalytic activity was evaluated by the discoloration of rhodamine B (RhB). It was indicated that, under visible light illumination, the photocatalytic rate constant of CdS/CeO2 was nearly 3.4 times and 28 times higher than that of pure CdS and pure CeO2, respectively, owing to the high oxygen storage capacity of the CdS/CeO2 heterostructure, and the inhibition of electron–hole pair recombination benefitting from efficient electron transfer from CdS NRs to CeO2 NPs. In addition, the degradation mechanism of RhB on CdS/CeO2 was also discussed. This durable nanocomposite catalyst, with its excellent combination of CdS NRs and CeO2 NPs, is able to be a promising catalyst for RhB degradation under visible-light irradiation.
1. Introduction
Photocatalytic degradation of pollutants through semiconductor compounds has become a promising approach to solve environmental problems during the last few decades. Among all semiconducting nanostructures, one-dimensional (1D) nanostructures have captured particular attention. At first, 1D geometry can result in a fast and long-distance electron transport. Moreover, their high length-to-diameter ratio can enhance the light absorption and scattering. At last, 1D structures own larger specific surface area that cannot be obtained in their bulk counterparts.1–4
Metal chalcogenides, such as CdS,5 ZnS,4 CuS,6 In2S3 (ref. 7) and SnS2,8 have been received growing attention on account of their suitable band gap which belongs to the visible spectrum. Among these, CdS, with a direct band gap of 2.4 eV, has been extensively investigated in numerous applications, such as gas sensors,9 lasing10 and waveguide material,11 solar cell,12 environment purification13 and hydrogen evolution,3,14 owing to its relatively high ability in harvesting visible light, favorable conduction band potential and inexpensive cost.14,15 However, the quick recombination of photo-generated charge carriers and the serious photocorrosion property are two major defects existing in the CdS catalyst.14 In order to solve these problems, different strategies included element doping,16 fabricating nano-scaled CdS of different geometric shapes (i.e. rings,10 belts,17 tubes18 and rods19) and sensitization with other metal oxides14,19 have been adopted. Particularly, heterostructures photocatalyst composed of CdS NRs and metal oxides opens up a new window to construct high-quality and high-efficiency visible-light-driven photocatalysts.14,19,20 Khan et al.14 reported a hierarchical three-dimensional NiO–CdS heteroarchitecture, which showed an ultra-high photocatalytic activity for hydrogen generation. The enhanced hydrogen production rate was mainly ascribed to the enlarged specific surface area and reduction of the recombination rate of electron–hole pairs. Moreover, 1D CdS/α-Fe2O3 nanostructures possessed higher activity than CdS NRs for photocatalytic degradation of methylene blue under visible-light illumination.13 On the other hand, earth metal oxides, such as La2O3,21 Pr6O11 (ref. 22) and CeO2,23 are usually chosen as co-catalysts to decorate other semiconductors for the fabrication of heterostructure photocatalysts. Among all, CeO2 is considered to be a promising catalyst for catalytic organics degradation benefiting from its UV absorbing ability, high electrical conductivity, extensively documented oxygen storage capacity,23 multiple redox states as well as extended stability.23–26 It is well established that CeO2 and its based composites have been explored in water dissociation,27 CO oxidation,28 pollutants degradation,29 hydrodenitrogenation30 and hydrogen production.31 Accordingly, sensitization titanium dioxide (TiO2) nanotube array with CeO2 NPs was conducted. The combination showed largely enhanced charge storage capacity owing to the increased number of surface active sites induced by the CeO2 NPs.25 To the best of our knowledge, there is no report concerning the synthesis of CeO2 nanoparticles sensitized CdS nanorods heterostructures photocatalyst for the application of dye degradation.
In this work, we demonstrated the photocatalytic degradation of rhodamine B under visible light illumination using a novel CdS nanorods/CeO2 nanoparticles heteroarchitecture fabricated by a facile hydrothermal route. The degradation mechanism of RhB dyes on CdS/CeO2 was proposed (Fig. 1).
 |
| | Fig. 1 Chemical structure of rhodamine B. | |
2. Experimental
For the sample preparation, characterization and photocatalytic test have been shown in the Experimental section in the ESI.†
3. Results and discussions
3.1. FE-SEM and TEM images
The detailed morphology and size of the synthesized products were visualized by SEM and TEM. As shown in Fig. 2a, the highly ordered CdS NRs displayed an average rod diameter of about 100 nm with a length of 1–2 μm. From Fig. 2b and c, it can be found that the CeO2 NPs with an average diameter around 12 nm were anchored on the wall of the CdS NRs. Acting as a “spacer”, the CeO2 NPs on CdS NRs can prevent the CdS NRs from aggregation.32 HRTEM measurements were further performed to investigate the detailed shape and crystal facets of the CdS/CeO2 heterostructure. As demonstrated in Fig. 2e, the different orientation and lattice fringe of CdS NRs and CeO2 NPs can be clearly observed. One interplanar distances of 0.34 nm can be indexed to the (002) direction of hexagonal CdS (d = 0.336 nm). The crystallite attached to the CdS with d-spacing of 0.315 nm matched well with the crystal facet (111) of cubic CeO2. The presence of integration interface between CdS (002) and CeO2 (111) further confirmed the strong interaction between CdS and CeO2 in CdS/CeO2.
 |
| | Fig. 2 (a) SEM images of CdS nanorods; (b) SEM image of CdS nanorods decorated with CeO2 nanoparticles; (c) TEM image of 1D CdS/CeO2 nanocomposites; (d) magnified TEM image of CdS/CeO2 heterostructure; (e) HRTEM images of the rectangular regions. | |
3.2. X-ray diffraction analysis
XRD patterns of the as-prepared CdS NRs, CeO2 NPs and CdS/CeO2 nanocomposite were shown in Fig. 3. It was observed that the phase of CdS/CeO2 samples was composited of cubic structure of CeO2 (JCPDS 43-1002) and hexagonal structure of CdS (JCPDS 41-1049). For CdS NRs, the characteristic peaks at 2θ values of 24.8°, 26.5°, 28.2°, 43.7°, 47.8° and 70.8° can be indexed to the (100), (002), (101), (110), (103) and (211) planes of hexagonal wurtzite crystal structured CdS NRs, respectively. The peaks of cubic fluorite structured CeO2 at 33.1°, 56.3° and 69.4° were originated from the (200), (311) and (400) diffractions, respectively. Though some crystal plane diffraction peaks of CeO2 may overlapped with the peaks of CdS, two main peaks {(200) and (311)} of CeO2 appeared in the XRD profiles of CdS/CeO2 confirmed the existence of CeO2 NPs on CdS NRs.
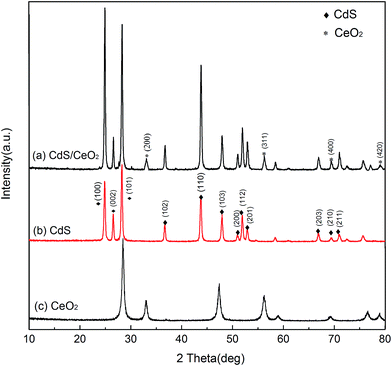 |
| | Fig. 3 XRD pattern of CdS/CeO2 nanocomposites (top); CdS nanorods (middle); CeO2 nanoparticles (bottom). | |
3.3. X-ray photoelectron spectroscopy (XPS) analysis
To investigate the chemical and oxidation states for Cd, S, Ce and O, XPS analysis was carried out. The spin–orbit components (3d5/2 and 3d3/2) of the Cd 3d peak were well deconvoluted for CdS/CeO2 nanocomposites by two curves at approximately 411.93 eV and 405.17 eV (Fig. 4a), manifesting that cadmium was in the Cd2+ state in the samples.15 Meanwhile, the determined binding energies of S 2p were found located at 161.51 eV and 162.7 eV (Fig. 4b), corresponding to sulfide species (S2−) of CdS/CeO2 composites.5 Fig. 4c showed the profile of high resolution XPS spectra of Ce 3d in CdS/CeO2 composites. It was found that a small amount of Ce3+ (885.4 and 904.1 eV) were coexisting with Ce4+ (882.1 eV, 888.9 eV, 898 eV, 900.3 eV, 907.6 eV and 916.6 eV) in the nanocomposites.28 By fitting the curves and calculating the Ce3+/(Ce3+ + Ce4+), a pronouncedly high Ce3+ ratio of 20.5% on CdS/CeO2 was identified, which was higher than that of CeO2/graphene (18.5%) reported in earlier study.33 Usually, Ce3+ content is proportional to oxygen vacancies, the ultra-high Ce3+ ratio denote that a certain amount of oxygen vacancies were introduced in the nanocomposites.34 This result was further confirmed by the XPS result of the O 1s region depicted in Fig. 4d. The O 1s spectrum was matched with three peaks at 529.12 eV, 531.94 eV and 533.68 eV. The distinct shoulder detected at 531.94 eV was corresponded to the adsorbed oxygen and weakly bonded oxygen species; the peak with the highest binding energy belonged to oxygen by hydroxide or adsorbed water on the catalyst surface. Both of the peaks illustrated above confirmed the existent of plenty of surface oxygen vacancies in CdS/CeO2. Meanwhile, the weakest peak, located at 529.12 eV, was found attributed to lattice oxygen in the anchored CeO2 nanocrystals.28,29,35 Generally, surface adsorbed oxygen is more reactive than lattice oxygen in oxidation reactions on account of its higher mobility.35 Hence, the higher surface adsorbed oxygen amount on CdS/CeO2 was helpful for the photocatalytic pollutant degradation, leading a significantly enhanced photocatalytic ability of CdS/CeO2 for the degradation of RhB.
 |
| | Fig. 4 High-resolution XPS spectrum of the (a) Cd 3d, (b) S 2p, (c) Ce 3d and (d) O 1s. | |
3.4. N2 adsorption–desorption
The specific surface areas were recorded from the N2 adsorption–desorption isotherms using the BET analysis method. Fig. 5 showed the nitrogen adsorption–desorption isotherms for pure CdS, CeO2 and CdS/CeO2 (RC = 20) compound. Apparent hysteresis loops can be distinguished on the adsorption–desorption isotherms of CdS nanorods, CeO2 nanoparticles and CdS/CeO2, indicating the existence of porous structures. The surface area data are summarized in Table 1. As we can see, the total pore volume of CdS/CeO2 is increased a lot, which may come from the voids between the aggregated CeO2 particles and CdS rods. Besides, the surface area increased by addition of CeO2 to CdS. For the pristine CdS nanorod, a specific surface area of 13.1 m2 g−1 was obtained, whereas for CdS/CeO2 sample (RC = 20), the specific surface area was 21.8 m2 g−1. The higher BET surface area for CdS/CeO2 nanocomposites was presumed to result from the CeO2 NPs (24 m2 g−1) linked on CdS NRs depressed the agglomeration of CdS NRs. Consequently, the increased surface area might be one of the reasons responsible for the enhanced photocatalytic activity.
 |
| | Fig. 5 N2 adsorption–desorption isotherms for CdS (the magenta line), CeO2 (the gray line) and CdS/CeO2 (the blue line). (Inset: the corresponding pore size distribution). | |
Table 1 Textural property of photocatalysts
| Materials |
BET surface area (m2 g−1) |
Total pore volume (m3 g−1) |
Average pore size (nm) |
| CdS |
13.121 |
0.026 |
3.178 |
| CeO2 |
24.087 |
0.029 |
4.726 |
| CdS/CeO2 |
21.764 |
0.068 |
3.465 |
3.5. UV-vis diffuse reflectance spectroscopy analysis
The Ultraviolet-visible (UV-vis) diffuse reflectance spectroscopy was employed to measure the optical properties of the samples. The corresponding absorption spectra of CdS, CeO2 and CdS/CeO2 (RC = 20) were displayed in Fig. 6. It was obvious that CdS/CeO2 nanocomposite exhibited a CdS absorption peak and a feature band edge of individual CeO2. Compared with pristine CdS and CeO2, CdS/CeO2 appeared spectral response in a broader visible-light region ascribed to the photosensitizing effect of CeO2. According to the following equation:where α is the absorption coefficient, ν is the light frequency, A is a constant, Eg is the band gap energy, and n is 1/2 and 2 for CdS and CeO2, respectively. The Eg of the as-prepared samples can be calculated from a plot depicting (αhν)n versus (hν) (shown in Fig. S2†). The extrapolated values (the straight line to the x axis) of (hν) at α = 0 gives absorption edge energies consistent with Eg = 2.37 eV and Eg = 2.95 eV for CdS and CeO2, respectively.
 |
| | Fig. 6 UV-vis DRS of CdS NRs, CeO2 NPs and CdS/CeO2 (RC = 20) heterostructures. | |
3.6. Photocatalytic activity
3.6.1. Effect of the amount of CeO2 nanoparticles loading on CdS nanorods. Decomposition of RhB (C0 = 40 mg L−1) under the identical conditions was carried out to determine the activities of samples with different molar ratio. The UV-vis spectral change of CdS–CeO2/RhB dispersion after 60 min adsorption in the dark was depicted in Fig. S3.† It was clear that no wavelength shift of the band at 554 nm was observed, indicating RhB was not decomposed during the dark reaction. The decreased absorbency of CdS–CeO2/RhB dispersion was corresponding to the extent of RhB adsorption on CdS/CeO2. The results of RhB degradation over CdS, CeO2 and CdS/CeO2 photocatalysts were shown in Fig. 7. It was noticeable that direct visible light irradiation of the RhB dye solution in the absence of catalysts was inconsequential toward degrading the dye. The photoactivity of the sample was strongly dependent on the RC value. With RC increasing from 0 to 20, the photoactivity of the sample for the RhB photodegradation was enhanced. The maximum degradation efficiency reached to 96.68% after 48 min for the sample (RC = 20). However, when RC further increased to 30, the photoactivity of the sample decreased instead. As expected, all samples showed higher photoactivities for the RhB degradation than that of pure CdS and CeO2 under visible light (λ ≥ 420 nm). Kinetics of the photocatalytic degradation of RhB was a pseudo-first-order,36 which can be explained in the Langmuir–Hinshelwood model:
 |
| | Fig. 7 Effect of CeO2 loading on photocatalytic degradation of RhB; the suspensions containing 0.4 g L−1 catalysts, natural pH = 4.3, initial concentration = 40 mg L−1, λ ≥ 420 nm. | |
The apparent rate constant (k) for the degradation of RhB on these samples were calculated by plotting ln(C0/Ct) versus t (Fig. 8), and the results were shown in Table 2. The excellent photocatalytic performances of CdS/CeO2 heterostructure for RhB degradation may be due to that (i) high oxygen storage capacity of CeO2 provided immense sources of active species; (ii) the strong interactions between CdS and CeO2 effectively prevented the photo-corrosion and leaching of CdS; and (iii) effective charge separation reduced the recombination rates of electron–hole pairs. The influence on RhB degradation was also investigated and displayed in the ESI.†
 |
| | Fig. 8 Kinetic fit for the degradation of RhB with pure CdS, CeO2 and CdS/CeO2 catalysts with different CeO2 contents. | |
Table 2 RhB degradation rates of photocatalysts
| Materials |
CdS |
CeO2 |
CdS/CeO2 (RC = 20) |
CdS/CeO2 (RC = 30) |
CdS/CeO2 (RC = 10) |
CdS/CeO2 (RC = 5) |
| Degradation rate (min−1) |
0.02089 |
0.00251 |
0.07003 |
0.04463 |
0.04182 |
0.03663 |
3.6.2. Analyses of intermediate products. The temporal evolution of the spectral changes during the photodegradation of RhB over CdS/CeO2 and CdS were given in Fig. 9a and b. It was noteworthy that original RhB depicted a major absorption band at 554 nm, and no shift of the adsorption band was observed after dark reaction for 1 h and visible light illumination for 48 min (Fig. S4†). In the presence of CdS, the absorption peak of RhB solution decreased slowly and the absorption band shifted from 554 to 548 nm within 48 min irradiation. By contrast, absorbance of the absorption peak at 554 nm decreased rapidly to almost zero and the wavelengths of the characteristic peaks shifted from 554 nm to 500 nm as the photodegradation reaction proceeded in the presence of CdS/CeO2. Obviously, hypsochromic shifts of the absorption maximum observed with CdS/CeO2 suspension were more pronounced than those observed with the CdS system. These hypsochromic shifts were assumed to result from the formation of a series of N-deethylated intermediates of RhB in a stepwise manner and the cleavage of the conjugated chromophore structure.21,37,38 To comprehend the photocatalytic degradation process better, intermediates of RhB during the photodegradation were further confirmed by HPLC monitoring equipped with a UV-visible diode array detector. The HPLC chromatogram of 0, 8, 16, 24, 32, 40 and 48 min filtrate of RhB/CdS–CeO2 (RC = 20) system were illustrated in Fig. 9c. The maximum concentration of RhB might appeared at the retention time of 4.42 min (peak a). Four other peaks (Fig. 9c, peak b–e), which may belong to N-deethylated intermediates, namely, N,N-diethyl-N′-ethylrhodamine (DER), N,N-diethylrhodamine (DR), N-ethyl-N′-ethylrhodamine (EER), N-ethylrhodamine (ER) or rhodamine (R) were also observed.37,38 With the quick diminishing of RhB, the concentration of other N-deethylated intermediates increased successively and subsequently decreased in succession with further irradiation, which revealed that the N-deethylation of RhB was a stepwise course. It was found that the first N-deethylated intermediate reached its maximum concentration after 16 min irradiation, whereas the final N-deethylated product got disappeared after 40 min irradiation. Based on these results, it was concluded that the RhB was effectively decomposed rather than merely bleached during the CdS/CeO2-assisted photodegradation. The fragmentation pathway of RhB photocatalytic degradation under visible light irradiation in CdS/CeO2 suspension was tentatively proposed as depicted in Fig. 10. Moreover, the total organic carbon (TOC) values of the solution at different time intervals during the RhB degradation was measured (Fig. S5†). After 48 min of irradiation, a relatively high mineralization rate of 86% of TOC removal was achieved, indicating that RhB has been mineralized efficiently under visible light irradiation.
 |
| | Fig. 9 Temporal spectral changes of RhB in aqueous (a) CeO2/CdS and (b) CdS dispersion under visible irradiation; (c) HPLC chromatograms of the N-deethylated intermediates at different irradiation intervals in the role of CdS/CeO2 (RC = 20). | |
 |
| | Fig. 10 Proposed pathway for the photocatalytic degradation of RhB dye under visible light. | |
3.6.3. Recycle test. To probe the practical reusability of the CdS/CeO2 catalyst in the decomposition of RhB, recycling tests were carried out by repeated reaction cycles under the same reaction condition. As shown in Fig. 11, the catalytic activity of CdS/CeO2 did not significantly decrease after six continuous cycles, in which the RhB removal efficiencies of CdS/CeO2 (RC = 20) were all above 90%. In sharp contrast, RhB removal efficiency of pure CdS for the first-cycle test was about 64%, and then obvious deactivation can be observed on CdS after four times recycles, indicating that the CdS/CeO2 owned a superior reusability in RhB degradation. To further demonstrate the stability of the catalyst, leaching of cadmium from CdS/CeO2 in solution after six cycle test was measured by via a Perkin-Elmer Analyst 700 atomic absorption spectrophotometer. For comparison, Cd content in solution after four cycle test for CdS catalyst was also detected. Results (inset of Fig. 11) indicated that the Cd ions released rapidly from CdS solid into the solution, which should be resulted from CdS photocorrosion. As for CdS/CeO2, rather low Cd species in CdS/CeO2 leached off even after being used six successive times. Moreover, there is no distinction between the XRD results of CdS/CeO2 samples before and after the reaction (Fig. S6†), suggesting its structural stability during the photocatalytic oxidation of the pollutant molecules. The improved stability of CdS/CeO2 may be ascribed to the intimate interfacial interaction39,40 between CdS and CeO2 and the introducing of high thermal stability CeO2.
 |
| | Fig. 11 Recycling test of CdS and CdS/CeO2 and the ICP analysis of residual Cd2+ in the used photocatalysts. | |
3.6.4. The proposed mechanism of RhB photodegradation. In order to determine the main active species responsible for the degradation of RhB over the optimum CdS/CeO2 composite (RC = 20), controlled photoactivity experiments were performed with adding different radical scavengers (0.1 mmol) at the natural pH of the dye. Fig. 12 reflected the photocatalytic activities of CdS/CeO2 (RC = 20) for the degradation of RhB in the presence of different radical scavengers, i.e., Ethylene Diamine Tetraacetic Acid (EDTA) scavenger for h+,41 isopropyl alcohol (IPA) for ˙OH and benzoquinone (BQ) for O2˙−, respectively. It was apparent that the order of affecting the rate for degradation of RhB followed that of O2˙− > h+ > ˙OH. It was shown that significant suppressing effect was obtained after addition of BQ (O2˙− quencher), suggesting the superoxide anion radical was the major oxidative species responsible for the photooxidative conversion of RhB.36,42 The band edge positions of the CB and VB of CdS and CeO2 was determined according the following equations:where X is the absolute electronegativity of the semiconductor (X is 5.18 eV and 5.57 eV for CdS and CeO2, respectively), Ee is the energy of free electrons on the hydrogen scale (4.5 eV) and Eg is the band gap of the semiconductor.26 The calculated CB and VB of CdS were −0.505 and 1.865 eV, respectively, which were more negative than those of CeO2 (−0.405 and 2.545 eV, respectively). The standard redox potential of O2/O2˙− and ˙OH/OH− is −0.33 and 2.38 V vs. NHE,42 respectively. That is, the CB potentials of CeO2 and CdS were both negative than the standard redox potential of O2/O2˙−, whereas the standard redox potential of ˙OH/OH− was positive to the VB of CdS. As a result, the photogenerated electrons on irradiated CdS/CeO2 heterostructure can reduce O2 to give O2˙−, while the photogenerated holes can hardly oxidize OH to give ˙OH, therefore O2˙− was the protagonist in RhB photodegradation by CdS/CeO2.
 |
| | Fig. 12 Effect of radical scavenger on the degradation of RhB using EDTA scavenger for h+, IPA for ˙OH and BQ for O2˙−, respectively. | |
Meanwhile, colorless organic molecules which do not absorb in visible regions were usually used for the evaluation of photocatalytic activity of photocatalyst.36,42–44 Therefore, colorless bisphenol A (BPA) and salicylic acid (SA) were chosen as the target pollutants. Prior to irradiation, the suspension was stirred for 1 h in dark to reach adsorption–desorption equilibrium. The sample was then irradiated with visible light with continuous stirring. After 1 h irradiation, the BPA and SA removal ratio of CdS/CeO2 (RC = 20) was 31.4% and 46.3%, respectively. While the pure CdS showed a degradation ratio of 5.2% and 7.7%, for BPA and SA, respectively. Therefore, it was suggested that the CdS/CeO2 composites is indeed a visible-light induced photocatalyst. Besides, considering the redox potential of E0RhB*/RhB˙+(−1.09 V vs. SHE) and E0RhB˙+/RhB (1.46 V vs. SHE),42 photo-induced electrons can transfer from photoexcited dye (RhB*) to the conduction bands of CdS, which has also been reported by Takuo et al.45,46 Based on the above discussion, a tentative illustration of possible interparticle electron transfer behavior can be proposed, as illustrated in Fig. 13. Under visible-light irradiation, photo-induced electrons easily transferred from photoexcited dye (RhB*) to the CB of CdS to form RhB+˙. These transferred electrons captured by adsorbed oxidants to generate active species (e.g., O2˙−). Afterwards, RhB+˙ could be consumed by these generated O2˙− radicals. On the other hand, the electrons could be photoexcited directly from the VB of CdS NRs to the CB of CdS by adsorbing light, forming electron–hole pairs. Immediately, the generated electrons of CdS transferred to the CB of CeO2. After that, the transferred electrons trapped by the adsorbed oxygen on the surface of the catalyst to produce reactive oxygen radicals (O2˙−). Finally, RhB dyes adsorbed on the surface of CdS/CeO2 was attacked by the activated oxygen to decolorize the solution. Simultaneously, holes generated in CdS/CeO2 can directly react with adsorbed dyes, making contributions to the RhB degradation process. The overall decomposition of RhB can be expressed by the following formula:
| | |
RhB∗ + CdS → RhB˙+ + CdS (e−)
| (6) |
| | |
CdS + hν → CdS (e− + h+)
| (7) |
| | |
CdS (e−) + CeO2 → CeO2 (e−)
| (8) |
| | |
e(CB,CeO2)− + O2 → O2˙−
| (9) |
| | |
RhB/RhB˙+ + (O2˙− + h+) → mineralized products → CO2 + H2O
| (10) |
 |
| | Fig. 13 Scheme diagram of the photocatalytic reaction of RhB on CdS/CeO2 heterostructure. | |
4. Conclusions
Nanocomposites made with CdS NRs and CeO2 NPs were successfully synthesized using a hydrothermal method. FE-SEM and TEM analyses showed that the CeO2 NPs were anchored on the wall of the CdS NRs strongly. XPS spectra and XRD result confirmed the formation of CdS/CeO2 heterostructures. The CeO2 played a vital role in achieving excellent photocatalytic performance of the synthesized composites toward degradation of RhB due to its large oxygen capacity and facilitation for the electron–hole pair separation. RhB was decomposed around 97% under visible light over CdS/CeO2, which was much enhanced than original CdS NRs (64%) and CeO2 NPs (12%). The sample (RC = 20) had the highest kinetic rate constant (0.07 min−1) owing to the effective charge separation and excellent redox capacity of photocatalysts. Results also demonstrated that the degradation efficiency increased with decreasing pH values. It is hoped that more ongoing research efforts could be paid to the earth metal oxides and the fabrication of 1D semiconductor nanostructures.
Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. 71431006, 21276069), the State Scholarship Fund and the Fundamental Research Funds for the Central Universities.
References
- S. Liu, N. Zhang, Z. R. Tang and Y. J. Xu, ACS Appl. Mater. Interface, 2012, 4, 6378–6385 CrossRef CAS PubMed.
- B. Weng, S. Liu, N. Zhang, Z. R. Tang and Y. J. Xu, J. Catal., 2014, 309, 146–155 CrossRef CAS PubMed.
- X. Wang, G. Liu, G. Q. Lu and H. M. Cheng, Int. J. Hydrogen Energy, 2010, 35, 8199–8205 CrossRef CAS PubMed.
- L. P. Wu, Y. L. Zhang, L. Z. Long, C. P. Cen and X. J. Li, RSC Adv., 2014, 4, 20716–20721 RSC.
- P. Gao, J. Liu, D. D. Sun and W. Ng, J. Hazard. Mater., 2013, 250–251, 412–420 CrossRef CAS PubMed.
- Y. Wang, L. Zhang, H. Jiu, N. Li and Y. Sun, Appl. Surf. Sci., 2014, 303, 54–60 CrossRef CAS PubMed.
- M. Xie, X. Dai, S. Meng, X. Fu and S. Chen, Chem. Eng. J., 2014, 245, 107–116 CrossRef CAS PubMed.
- X. Yuan, H. Wang, Y. Wu, X. Chen, G. Zeng, L. Leng and C. Zhang, Catal. Commun., 2015, 61, 62–66 CrossRef CAS PubMed.
- L. Zhu, C. Feng, F. Li, D. Zhang, C. Li, Y. Wang, Y. Lin, S. Ruan and Z. Chen, RSC Adv., 2014, 4, 61691–61697 RSC.
- Z. Hu, X. Guo and L. Tong, Appl. Phys. Lett., 2013, 103, 183104 CrossRef PubMed.
- A. Pan, D. Liu, R. Liu, F. Wang, X. Zhu and B. Zou, Small, 2005, 1, 980–983 CrossRef CAS PubMed.
- R. Yang, D. Wang, L. Wan and D. Wang, RSC Adv., 2014, 4, 22162–22171 RSC.
- Le Wang, H. Wei, Y. Fan, X. Gu and J. Zhan, J. Phys. Chem. C, 2009, 113, 14119–14125 CAS.
- Z. Khan, M. Khannam, N. Vinothkumar, M. De and M. Qureshi, J. Mater. Chem., 2012, 22, 12090–12095 RSC.
- R. C. Pawar and C. S. Lee, Appl. Catal., B, 2014, 144, 57–65 CrossRef CAS PubMed.
- J. E. C. Aioliva, R. Patiño and A. I. Oliva-Avilés, Bull. Mater. Sci., 2014, 37, 247–255 CrossRef.
- Y. Ye, L. Dai, X. Wen, P. Wu, R. Pen and G. Qin, ACS Appl. Mater. Interface, 2010, 2, 2724–2727 CrossRef CAS.
- Y. Xiong, Y. Xie, J. Yang, R. Zhang, C. Wu and G. Du, J. Mater. Chem., 2002, 12, 3712–3716 RSC.
- C. Luan, T. L. Wong and J. A. Zapien, J. Cryst. Growth, 2013, 374, 65–70 CrossRef CAS PubMed.
- S. Yan, D. Hu, J. Wu, X. Xu, J. Wang and Z. Xiao, J. Alloys Compd., 2011, 509, L239–L243 CrossRef CAS PubMed.
- H. Xu, H. Li, G. Sun, J. Xia, C. Wu, Z. Ye and Q. Zhang, Chem. Eng. J., 2010, 160, 33–41 CrossRef CAS PubMed.
- C. Karunakaran, R. Dhanalakshmi and P. Anilkumar, Chem. Eng. J., 2009, 151, 46–50 CrossRef CAS PubMed.
- N. Laosiripojana, W. Sutthisripok and S. Assabumrungrat, Chem. Eng. J., 2007, 127, 31–38 CrossRef CAS PubMed.
- Q. Dai, J. Wang, J. Yu, J. Chen and J. Chen, Appl. Catal., B, 2014, 144, 686–693 CrossRef CAS PubMed.
- H. Wen, Z. Liu, Q. Yang, Y. Li and J. Yu, Electrochim. Acta, 2011, 56, 2914–2918 CrossRef CAS PubMed.
- Z.-M. Yang, G.-F. Huang, W.-Q. Huang, J.-M. Wei, X.-G. Yan, Y.-Y. Liu, C. Jiao, Z. Wan and A. Pan, J. Mater. Chem. A, 2014, 2, 1750 CAS.
- D. R. Mullins, P. M. Albrecht, T.-L. Chen, F. C. Calaza, M. D. Biegalski, H. M. Christen and S. H. Overbury, J. Phys. Chem. C, 2012, 116, 19419–19428 CAS.
- Z. Wang, Q. Wang, Y. Liao, G. Shen, X. Gong, N. Han, H. Liu and Y. Chen, ChemPhysChem, 2011, 12, 2763–2770 CrossRef CAS PubMed.
- Y. G. Wang, B. Li, L. F. Cui, S. F. Kang, X. Li and L. H. Zhou, Appl. Catal., B, 2013, 130–131, 277–284 CrossRef CAS PubMed.
- Z. Sun, X. Li, A. Wang, Y. Wang and Y. Chen, Top. Catal., 2012, 55, 1010–1021 CrossRef CAS.
- X. H. Lu, S. L. Xie, T. Zhai, Y. F. Zhao, P. Zhang, Y. L. Zhang and Y. X. Tong, RSC Adv., 2011, 1, 1207–1210 RSC.
- Z. Ji, X. Shen, M. Li, H. Zhou, G. Zhu and K. Chen, Nanotechnology, 2013, 24, 115603 CrossRef PubMed.
- L. Jiang, M. Yao, B. Liu, Q. Li, R. Liu, H. Lv, S. Lu, C. Gong, B. Zou, T. Cui, B. Liu, G. Hu and T. Wågberg, J. Phys. Chem. C, 2012, 116, 11741–11745 CAS.
- M. Yu, Y.-A. Zhu, Y. Lu, G. Tong, K. Zhu and X. Zhou, Appl. Catal., B, 2015, 165, 43–56 CrossRef CAS PubMed.
- W. Shan, F. Liu, H. He, X. Shi and C. Zhang, Catal. Today, 2012, 184, 160–165 CrossRef CAS PubMed.
- C. S. Pan, H. B. Fu, W. Q. Yao and Y. F. Zhu, J. Phys. Chem. B, 2005, 109, 22432–22439 CrossRef PubMed.
- S. Y. Kai Yu, H. He, C. Sun, C. Gu and Y. Ju, J. Phys. Chem. A, 2009, 113, 10024–10032 CrossRef PubMed.
- T. S. Natarajan, M. Thomas, K. Natarajan, H. C. Bajaj and R. J. Tayade, Chem. Eng. J., 2011, 169, 126–134 CrossRef CAS PubMed.
- X. Gao, H. B. Wu, L. Zheng, Y. Zhong, Y. Hu and X. W. Lou, Angew. Chem., Int. Ed., 2014, 53, 5917–5921 CrossRef CAS PubMed.
- H. Li, X. He, Z. Kang, H. Huang, Y. Liu, J. Liu, S. Lian, C. H. Tsang, X. Yang and S. T. Lee, Angew. Chem., Int. Ed., 2010, 49, 4430–4434 CrossRef CAS PubMed.
- Y. Liu, X. Yuan, H. Wang, X. Chen, S. Gu, Q. Jiang, Z. Wu, L. Jiang and G. Zeng, RSC Adv., 2015, 5, 33696–33704 RSC.
- W. Li, D. Li, S. Meng, W. Chen, X. Fu and Y. Shao, Environ. Sci. Technol., 2011, 45, 2987–2993 CrossRef CAS PubMed.
- M. Zhang, C. Chen, W. Ma and J. Zhao, Angew. Chem., 2008, 47, 9730–9733 CrossRef CAS PubMed.
- J. Sun, G. Chen, J. Wu, H. Dong and G. Xiong, Appl. Catal., B, 2013, 132–133, 304–314 CrossRef CAS PubMed.
- T. Watanabe, T. Tadashi and K. Honda, J. Phys. Chem., 1977, 81, 1845–1851 CrossRef CAS.
- T. Takirawa, T. Watanabe and K. Honda, J. Phys. Chem., 1978, 82, 1391–1396 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra16114b |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 












