
The effect of liquid stabilization on the structures and the conductive properties of polyimide-based graphite fibers
Abstract
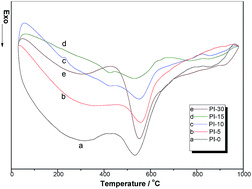
Polyimide (PI) fibers, stabilized under reflux of hot nitric acid, are carbonized and graphitized in an inert atmosphere to obtain polyimide-based graphite fibers (PI-GFs). The results of DSC show that the exothermic interval of stabilized PI fibers becomes broad, accompanied by a reduction in total heat release, from 875.18 mW mg−1 (non-stabilized) to 166.13 mW mg−1 (stabilized for 15 min). And the carbon yields substantially increase, from the initial 15.98% (non-stabilized) to 49.93% (stabilized for 30 min) as proved by TGA. In addition, when the stabilization time is over 15 min, some defects such as cracks will appear on the fiber surface, which have a negative influence on the properties of the resulting PI-GFs. After being graphitized at 2800 °C, the stabilized PI-GFs have a high degree of graphitization and thermal conductivity which could be as high as 415.35 W m−1 K−1. It is indicated that PI fibers may be a good potential candidate for graphite fibers with high conductivity.
 Please wait while we load your content...
Please wait while we load your content...

