
Penchinones A–D, two pairs of cis-trans isomers with rearranged neolignane carbon skeletons from Penthorum chinense†
Ya-Cong Heab,
Cheng Peng*ab,
Xiao-Fang Xieab,
Ming-Hua Chenc,
Xiao-Nian Lid,
Meng-Ting Liab,
Qin-Mei Zhouab,
Li Guoab and
Liang Xiong*ab
aState Key Laboratory Breeding Base of Systematic Research, Development and Utilization of Chinese Medicine Resources, Chengdu University of Traditional Chinese Medicine, Chengdu 611137, China. E-mail: pengchengchengdu@126.com; xiling0505@126.com
bSchool of Pharmacy, Chengdu University of Traditional Chinese Medicine, Chengdu 611137, China
cInstitute of Medicinal Biotechnology, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100050, China
dState Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, China
First published on 4th September 2015
Abstract
Penchinones A–D (1–4), two pairs of novel cis-trans lignan isomers with rearranged neolignane carbon skeletons, together with six known flavonoids (5–10), were isolated from the ethyl acetate-soluble portion of a hepatoprotective water decoction of Penthorum chinense. Their structures were determined by extensive spectroscopic data analysis, ECD calculation, and single-crystal X-ray diffraction. Penchinones C and D (3 and 4) featured an unprecedented 7,3′-neolignane carbon skeleton. Penchinone A (1) exhibited protective activity against acetaminophen-induced damage to the HL7702 hepatocyte and selective cytotoxicity against the human cancer ovarian cell line, Hey, in vitro.
Introduction
Lignans are a class of naturally occurring phenylpropanoid dimers that may play an important role in plant growth and plant defense against various biological pathogens and pests.1 In addition to their biological functions in plants, lignans have significant pharmacological effects, including antibacterial, antiviral, antitumor, antioxidant, anti-inflammatory, immunosuppressive, cardiovascular, and hepatoprotective activities.1–3 Although the molecular backbone consists only of two phenylpropane (C6–C3) units, lignans exhibit an enormous range of structural diversity because of the various linkage patterns of their C6–C3 units. Under IUPAC nomenclature, lignans are classified into three types: classical lignans, neolignans, and oxyneolignans. If the two C6–C3 units are linked by a direct carbon–carbon bond other than an 8-8′ linkage, the compounds are defined as neolignans.4 In the past few decades, phytochemical investigations have led to the isolation of hundreds of neolignans of 21 subtypes: 8,3′-neolignane, 3,3′-neolignane, 8,1′-neolignane, 1,3′-neolignane, 1,7′-neolignane, 8,7′-neolignane, 2,9′-neolignane, 9,9′-neolignane, 8,9′-neolignane, 3,4′-neolignane, 4,4′-neolignane, 5′,7-cyclo-8,3′-neolignane, 1′,7-cyclo-8,3′-neolignane, 6′,9-cyclo-8,3′-neolignane, 7,9′-cyclo-8,1′-neolignane, 3′,7-cyclo-8,1′-neolignane, 2,7′-cyclo-8,7′-neolignane, 7,8′-cyclo-8,7′-neolignane, 2,6′-cyclo-1,3′-neolignane, 1′,7:2′,8-dicyclo-8,3′-neolignane, and 1′,3:2,2′:6,6′-tricyclo-1,3′-neolignane.2–4Penthorum, a genus originally in the family Crassulaceae, was moved to the family Saxifragaceae in 1981, but has been considered a separate family by the APG (Angiosperm Phylogeny Group) system since 1998. Penthorum chinense Pursh is one of the only two species in the family Penthoraceae, and is widely distributed and cultivated in Sichuan Province, China.5 In this region, the plant is commonly used as a folk medicine for the treatment of hepatic and gall diseases, and as an ingredient of various comestibles.6,7 Our previous investigation of the hepatoprotective portion of the water decoction of P. chinense led to the identification of seven lignans. Some exhibited protective activities against acetaminophen-induced hepatocyte injury.7 As continuous search for hepatoprotective secondary metabolites from P. chinense, the bioactive portion was further studied, affording two pairs of cis-trans isomers with unprecedented rearranged neolignane skeletons (1–4) and six known flavonoids (5–10) (Fig. 1). We present herein the details of isolation, structure elucidation, hypothetical biogenesis, and bioactivities of 1–4.
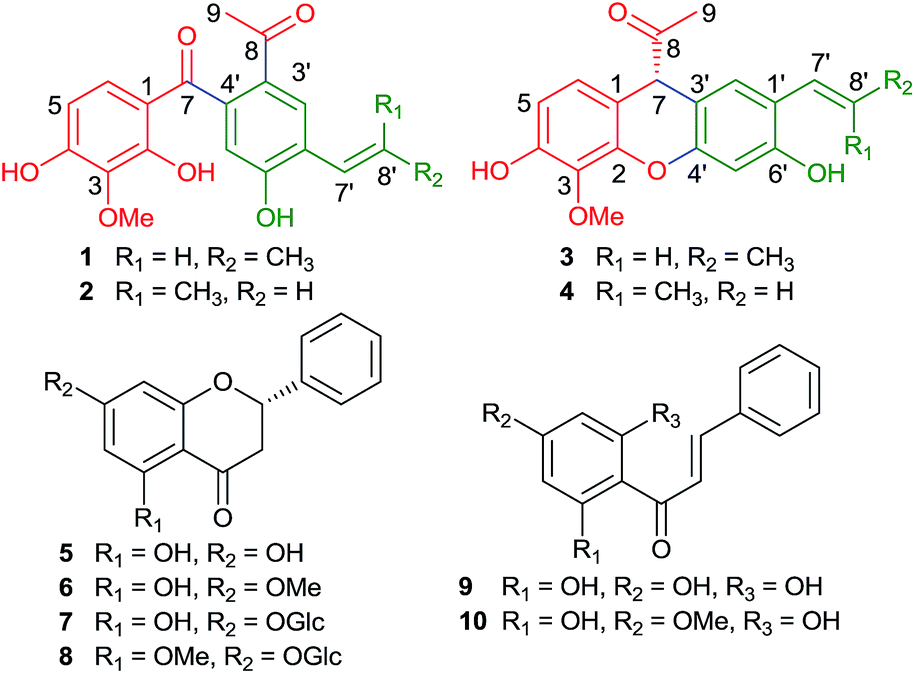 | ||
| Fig. 1 Structures of compounds 1–10. | ||
Results and discussion
Penchinone A (1) exhibited IR absorptions characteristic of hydroxyl (3317 cm−1), carbonyl (1652 and 1614 cm−1), aromatic ring and olefinic bond (1580, 1503, and 1433 cm−1) functionalities. The molecular formula of 1 was C19H18O6, with 11 degrees of unsaturation, as indicated by HRESIMS (m/z 365.1002 [M + Na]+, calcd 365.1001). The 1H NMR spectrum of 1 exhibited resonances attributable to a 1,2,3,4-tetrasubstituted phenyl [δH 6.27 (d, J = 8.4 Hz) and 6.72 (d, J = 8.4 Hz)], a 1,2,4,5-tetrasubstituted phenyl [δH 8.05 (s) and 6.68 (s)], a trans-propenyl [δH 6.72 (dq, J = 15.6, 1.2 Hz), 6.50 (dq, J = 15.6, 6.6 Hz), and 1.94 (dd, J = 6.6, 1.2 Hz)], and an aromatic acetyl group (δH 2.50), together with an aromatic methoxy (δH 3.88) (Table 1). The 13C NMR and DEPT spectra of 1 revealed 19 carbon resonances assignable to the above protonated units and 10 quaternary carbons, including two ketone carbonyl (δC 203.8 and 199.0) and eight aromatic quaternary carbons (four of which are oxygenated).| No. | 1 | 2 | 3 | 4 | ||||
|---|---|---|---|---|---|---|---|---|
| δH | δC | δH | δC | δH | δC | δH | δC | |
| a NMR data (δ) were measured at 600 MHz for 1H and at 150 MHz for 13C in CD3OD. Proton coupling constants (J) in Hz are given in parentheses. The assignments were based on 1H–1H gCOSY, gHSQC, and gHMBC experiments. | ||||||||
| 1 | 115.3 | 115.5 | 112.2 | 112.2 | ||||
| 2 | 158.6 | 158.3 | 146.1 | 146.1 | ||||
| 3 | 136.1 | 136.1 | 137.3 | 137.3 | ||||
| 4 | 158.7 | 158.5 | 151.9 | 151.9 | ||||
| 5 | 6.27 d (8.4) | 109.0 | 6.29 d (9.0) | 108.9 | 6.61 d (8.4) | 112.5 | 6.62 d (8.4) | 112.5 |
| 6 | 6.72 d (8.4) | 130.0 | 6.74 d (9.0) | 129.9 | 6.76 d (8.4) | 124.5 | 6.76 d (8.4) | 124.5 |
| 7 | 203.8 | 203.8 | 4.69 s | 54.2 | 4.70 s | 54.1 | ||
| 8 | 199.0 | 198.7 | 208.7 | 208.6 | ||||
| 9 | 2.50 s | 26.8 | 2.48 s | 26.8 | 1.88 s | 25.4 | 1.90 s | 25.5 |
| 1′ | 127.4 | 126.6 | 123.4 | 122.4 | ||||
| 2′ | 8.05 s | 130.7 | 7.92 s | 134.2 | 7.13 s | 127.4 | 7.01 s | 130.9 |
| 3′ | 128.8 | 128.1 | 110.8 | 110.1 | ||||
| 4′ | 140.8 | 141.3 | 151.2 | 151.3 | ||||
| 5′ | 6.68 s | 116.1 | 6.74 s | 115.8 | 6.63 s | 104.3 | 6.67 s | 104.2 |
| 6′ | 159.9 | 160.4 | 156.4 | 157.1 | ||||
| 7′ | 6.72 dq (15.6, 1.2) | 126.0 | 6.54 dq (11.4, 1.8) | 125.0 | 6.56 brd (15.6) | 125.8 | 6.44 dq (12.0, 1.8) | 125.6 |
| 8′ | 6.50 dq (15.6, 6.6) | 129.2 | 5.96 dq (11.4, 7.2) | 129.5 | 6.17 dq (15.6, 6.6) | 126.4 | 5.77 dq (12.0, 6.6) | 127.1 |
| 9′ | 1.94 dd (6.6, 1.2) | 19.1 | 1.90 dd (7.2, 1.8) | 15.0 | 1.86 dd (6.6, 1.2) | 19.0 | 1.80 dd (6.6, 1.8) | 14.8 |
| OMe | 3.88 s | 60.8 | 3.89 s | 60.9 | 3.92 s | 61.6 | 3.93 s | 61.7 |
To determine the structure of 1, a comprehensive analysis of the 2D NMR data was conducted. In the 1H–1H gCOSY spectrum of 1, the homonuclear coupling correlations between H-5/H-6 and H-7′/H-8′/H3-9′ verified the presence of both the 1,2,3,4-tetrasubstituted phenyl and the propenyl (Fig. 2). The connection of the two phenyls via the carbonyl C-7 could be deduced from the key HMBC correlations of H-6 and H-5′ with C-7, together with the weak four-bond W-type correlations of H-5 and H-2′ with C-7. The acetyl group was assigned to C-3′, based on the HMBC correlations from H-2′ to C-8, H3-9 to C-8/C-3′, and H-5′ to C-3′. Meanwhile, the HMBC cross peaks of H-7′ with C-2′/C-6′/C-9′ and H-8′ with C-1′ revealed that the trans-propenyl was located at C-1′. Furthermore, the HMBC correlations of H-5 with C-1/C-3/C-4, H-6 with C-2/C-4, OMe with C-3, H-2′ with C-4′/C-6′/C-7′, and H-5′ with C-1′/C-3′, together with their chemical shifts and molecular formula, indicated that the methoxyl and three hydroxyl groups were substituted at C-3, C-2, C-4, and C-6′, respectively. Thus, the structure of 1 was established.
 | ||
| Fig. 2 The key 1H–1H COSY and HMBC correlations of 1. | ||
The spectroscopic data of penchinone B (2) indicated that it was an analog of 1 with the same molecular formula: C19H18O6. A comparison of the NMR data of 2 and 1 revealed a cis-propenyl in 2, replacing the trans-propenyl unit in 1 (Table 1). This was confirmed by 2D NMR data analysis. Since some weak four- and five-bond correlations could also be found in the HMBC spectrum, the structure of 2 was further confirmed using single-crystal X-ray crystallographic analysis. An ORTEP drawing, with the atom numbering indicated, is shown in Fig. 3.
 | ||
| Fig. 3 ORTEP drawing for 2. | ||
Penchinone C (3) also exhibited spectroscopic resonances for a 1,2,3,4-tetrasubstituted phenyl, a 1,2,4,5-tetrasubstituted phenyl, a trans-propenyl, and an acetyl, similar to those of 1. However, the 1H NMR, 13C NMR, and DEPT spectra revealed that an obviously deshielded isolated methine (δH 4.69, δC 54.2) in 3 replaced the ketone carbonyl in 1. The HRESIMS data for 3 indicated that it had a molecular formula of C19H18O5. These data suggested that 3 was a lignane with two C6–C3 units, which was confirmed by the 2D NMR experiments. In the HMBC spectrum, correlations from H-5 to C-1/C-3/C-4, H-6 to C-2/C-4/C-7, and OMe to C-3, verified the presence of the 1,2,3,4-tetrasubstituted phenyl group with a methoxy unit at C-3 and two oxygenated substituents at C-2 and C-4. The HMBC correlations of H-2′ with C-4′/C-6′/C-7′, H-5′ with C-1′/C-3′/C-4′/C-6′, H-8′ with C-1′/C-9′, and H3-9′ with C-7′/C-8′, combined with the relevant 13C NMR data and the 1H–1H COSY correlations of H-7′/H-8′/H3-9′, indicated that the trans-propenyl and two oxygenated substituents were attached to C-1′, C-4′, and C-6′ of the 1,2,4,5-tetrasubstituted phenyl, respectively. The connection of the two phenyls via C-1–C-7–C-3′ bonds was revealed by the HMBC correlations from H-7 to C-1, C-2, C-6, C-2′, C-3′, and C-4′. The acetyl group was further linked to C-7 by HMBC correlations from H-7 to C-8/C-9, and H3-9 to C-7/C-8. In accordance with the molecular formula and the degrees of unsaturation of 3, the remaining four oxygenated substituents (C-2, C-4, C-4′, and C-6′) could be conjectured to comprise an oxygen bridge connecting C-2 with C-4′, and two hydroxyls at C-4 and C-6′.8 This was confirmed by their chemical shifts [δC 146.1 (C-2), 151.9 (C-4), 151.2 (C-4′), and 156.4 (C-6′)], even though no long-range HMBC evidence for the linkage was provided. Thus, compound 3 was determined to have the following structure: (7′E)-4,6′-dihydroxy-3-methoxy-2,4′-epoxy-7,3′-neolignan-7′-ene-8-one.
The absolute configuration of C-7 in 3 was determined via an ECD (electronic circular dichroism) calculation using the time-dependent density functional theory (TDDFT) method.9,10 As shown in Fig. 4, the theoretically calculated ECD spectra of (R)-3 was in agreement with the experimental ECD spectra of 3, revealing the 7R configuration of 3.
 | ||
| Fig. 4 The experimental ECD spectra of 3 (blue) and the calculated ECD spectra of (R)-3 (red) and (S)-3 (black). | ||
Penchinone D (4) has the same molecular formula as that of 3, as indicated by the HRESIMS analysis. The 1D NMR data (Table 1) were similar to those of 3, with the exception of the substitution of the resonances for the trans-propenyl moiety in 3 by those attributable to a cis-propenyl unit in 4 [δH 6.44 (dq, J = 12.0, 1.8 Hz), 5.77 (dq, J = 12.0, 6.6 Hz), and 1.80 (dd, J = 6.6, 1.8 Hz); δC 125.6 (C-7′), 127.1 (C-8′), and 14.8 (C-9′)]. The 2D NMR data and [α]D value of 4 confirmed that it was 7′Z-isomer of 3.
The biosynthetic precursors of 1–4 were proposed to be penthorin A (11) and B (12), as the two compounds were previously isolated from the same extract of P. chinense.7 In addition, their analogs have been detected in this species by other researchers.8 As shown in Scheme 1, penthorin A and B are enolized first, after which two possible biosynthetic pathways (routes I and II) are postulated for 1/2 and 3/4, respectively. In route I, the oxidation of the enol intermediate would open the 7,8-bond, liberating a carboxyl group at C-1 and an acetyl group at C-3′. Then, an enzyme-catalyzed reduction reaction occurs to produce an aldehyde intermediate. Finally, we determined that the key biochemical reaction would be a free radical reaction combined with a proton transfer, which reconstructs a C-7/C-4′ carbon bond to produce 1/2. As indicated in route II, the protonation of 7-OH in the enol unit and the loss of H2O generate a carbocation center at C-7. Subsequently, an enzymatic rearrangement could lead to the migration of the 8,3′-bond to C-7. Then, the carbocation center created at C-8 would be neutralized by H2O, with the subsequent loss of a proton forming an enol intermediate and producing 3/4.
 | ||
| Scheme 1 Hypothetical biogenetic pathway for 1–4. | ||
The six known compounds were identified by comparison of spectroscopic data with those reported in the literatures as pinocembrin (5),11 pinostrobin (6),12 pinocembrin-7-O-β-D-glucopyranoside (7),13 5-methoxy-pinocembrin-7-O-β-D-glucoside (8),14 (E)-3-phenyl-1-(2,4,6-trihydroxyphenyl)prop-2-en-1-one (9),15 and pinostrobin chalcone (10).16
Based on the linking position of the two C6–C3 units, the reported neolignans were divided into 21 carbon skeletons (Fig. 5). The majority of the neolignans were 8,3′-neolignanes, 8,1′-neolignanes, and 3,3′-neolignanes, especially 4′,7-epoxy-8,3′-neolignanes (benzofurans).3 In recent years, more than one hundred 3′,7-cyclo-8,1′-neolignanes, 5′,7-cyclo-8,3′-neolignanes, and 7,8′-cyclo-8,7′-neolignanes have been also detected in several families, including the Lauraceae,17,18 Piperaceae,19,20 Magnoliaceae,21 and Bignoniaceae.22 However, other types of neolignanes are comparatively rare, according to the literature of the past few decades.23–27 This study discovered two unprecedented rearranged neolignane carbon skeletons from the plant kingdom.
 | ||
| Fig. 5 The reported carbon skeletons of neolignanes. | ||
Since new compounds 1–4 were isolated from the hepatoprotective water decoction of P. chinense, they were evaluated further to determine their protective activities against acetaminophen (AP)-induced damage to HL7702 hepatocytes in vitro, with the exception of compound 4, because the available sample quantity was too small to conduct the analysis. Compounds 2 and 3 exhibited weak activity, while a significant increase in the viability of HL7702 cells examined via a MTT colorimetric assay showed that 1 provided effective protection against AP-induced damage (Fig. 6). Furthermore, Image-iT® DEAD Green™ viability stain and Hoechst 33342 staining were used to demonstrate that 1 could protect damaged HL7702 cells, using a high-content screening (HCS) experiment (Fig. 7). The survival rate of acetaminophen-treated HL7702 cells rose from 35.7% in the model group to 91.0% and 86.4% in two test groups containing 1 at 10 μM and 1 μM, respectively. Interestingly, in the assay of cytotoxic activity against human hepatocellular carcinoma cell line HePG-2, lung cancer cell lines A549 and NCI-H1975, breast cancer cell lines MDA-MB-231 and MCF-7, and ovarian cancer cell line Hey, compound 1 also exhibited selective cytotoxicity against Hey with IC50 value of 13.7 μg mL−1.
 | ||
| Fig. 6 Protective activity of 1 against AP-induced HL7702 cell damage (#p < 0.05 vs. control, *p < 0.05 vs. model). | ||
 | ||
| Fig. 7 Nuclear DNA staining of the HL7702 cells, where Hoechst 33342 stained live and dead cells blue, Image-iT® DEAD Green™ viability stain stained dead cells green. | ||
Experimental
General experimental procedure
Optical rotations were measured using a Perkin-Elmer 341 plus. CD spectra were recorded on a JASCO J-815 CD spectrometer. IR spectra were recorded on a Nicolet 5700 FT-IR microscope instrument. NMR spectra were obtained using a Bruker-AVIIIHD-600 spectrometer with the solvent peaks used as the references. HRESIMS spectra were measured using a Waters Synapt G2 HDMS. Column chromatography (CC) was performed using silica gel (200–300 mesh, Yantai Institute of Chemical Technology, Yantai, China), MCI gel CHP 20P (75–150 μm, Mitsubishi Chemical, Co., Japan), and Sephadex LH-20 (Amersham Pharmacia Biotech AB, Uppsala, Sweden). HPLC separation was performed using an instrument equipped with a Cometro 6000LDS pump, a Cometro 6000PVW UV/VIS detector, and an Ultimate (250 × 10 mm2) preparative column packed with C18 (5 μm). TLC was performed using glass precoated silica gel GF254 plates (Qingdao Marine Chemical Inc., Qingdao, China).Plant material
P. chinense was collected in July of 2012 from the culture field in Guling, Sichuan Province, China. Plant identity was verified by Prof. Min Li (Chengdu University of TCM, Sichuan, China). A voucher specimen (SGHC-20120725) was deposited at the School of Pharmacy, Chengdu University of TCM, Chengdu, China.Extraction and isolation
The air-dried plant material (16 kg) was decocted with H2O (130 L; 3 × 1 h). The aqueous extracts were combined and evaporated under reduced pressure to yield a dark-brown residue (1.8 kg), which was suspended in H2O (2.0 L) and then successively partitioned with EtOAc (8 × 2.0 L) and n-BuOH (8 × 2.0 L). The EtOAc extract (470 g) was chromatographed over a D-101 macroporous adsorbent resin column. Successive elution of the column with 30% EtOH, 50% EtOH, 75% EtOH, and 95% EtOH (15 L each) yielded four portions. The third portion (65 g) eluted by 75% EtOH was subjected to silica gel CC using a gradient elution of increasing MeOH (0–100%) in CH2Cl2 to give 11 fractions (F1 − F11). F3 was separated over the Sephadex LH-20 column eluted with petroleum ether–CHCl3–MeOH (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5:1) to afford five subfractions (F3-1 − F3-5). The subfraction F3-3 was subjected to silica gel CC using a gradient elution of increasing MeOH (0–100%) in CH2Cl2 to give six parts (F3-3-1 − F3-3-6). Further purification of F3-3-3 with preparative TLC (CHCl3–MeOH, 30:1) followed by reversed-phase semipreparative HPLC (68% MeOH in H2O) yielded 3 (2.7 mg) and 4 (0.7 mg). F3-3-4 was further separated on the preparative TLC (CHCl3–MeOH, 40:1) and reversed-phase semipreparative HPLC (80% MeOH in H2O) to afford 1 (7.5 mg) and 2 (11.8 mg).
:5:1) to afford five subfractions (F3-1 − F3-5). The subfraction F3-3 was subjected to silica gel CC using a gradient elution of increasing MeOH (0–100%) in CH2Cl2 to give six parts (F3-3-1 − F3-3-6). Further purification of F3-3-3 with preparative TLC (CHCl3–MeOH, 30:1) followed by reversed-phase semipreparative HPLC (68% MeOH in H2O) yielded 3 (2.7 mg) and 4 (0.7 mg). F3-3-4 was further separated on the preparative TLC (CHCl3–MeOH, 40:1) and reversed-phase semipreparative HPLC (80% MeOH in H2O) to afford 1 (7.5 mg) and 2 (11.8 mg).
Physical-chemical properties of 1–4
ε) 214 (4.05), 252 (4.23), 289 (4.11) nm; IR (KBr) νmax 3318, 2941, 1652, 1614, 1581, 1503, 1433, 1359, 1301, 1159, 1091, 1043, 969, 814, 794 cm−1; 1H NMR (MeOH-d4, 600 MHz) data and 13C NMR (MeOH-d4, 150 MHz) data, see Table 1. (+)-HR-ESIMS m/z 365.1002 [M + Na]+ (calcd for C19H18O6Na, 365.1001).ε) 215 (4.14), 243 (4.21), 288 (4.17) nm; IR (KBr) νmax 3295, 2974, 2936, 1653, 1602, 1568, 1504, 1427, 1362, 1278, 1161, 1092, 1044, 971, 812, 792, 757 cm−1; 1H NMR (MeOH-d4, 600 MHz) data and 13C NMR (MeOH-d4, 150 MHz) data, see Table 1. (+)-HR-ESIMS m/z 365.1003 [M + Na]+ (calcd for C19H18O6Na, 365.1001).ε) 224 (4.33), 268 (3.96), 308 (3.53) nm; CD (MeCN) 244 (Δε − 1.46), 290 (Δε + 0.64) nm; IR (KBr) νmax 3364, 2927, 2853, 1706, 1600, 1580, 1497, 1460, 1314, 1262, 1183, 1108, 1072, 802 cm−1; 1H NMR (MeOH-d4, 600 MHz) data and 13C NMR (MeOH-d4, 150 MHz) data, see Table 1. (−)-HR-ESIMS m/z 325.1075 [M − H]− (calcd for C19H17O5, 325.1076).ε) 222 (4.32), 259 (3.98), 301 (3.55) nm; 1H NMR (MeOH-d4, 600 MHz) data and 13C NMR (MeOH-d4, 150 MHz) data, see Table 1. (−)-HR-ESIMS m/z 325.1073 [M − H]− (calcd for C19H17O5, 325.1076).X-ray crystallography of compound 2
C19H20O7, M = 360.35, orthorhombic, a = 9.9508(3) Å, b = 19.0262(6) Å, c = 19.3662(6) Å, α = 90.00°, β = 90.00°, γ = 90.00°, V = 3666.5(2) Å3, T = 100(2) K, space group Pbca, Z = 8, μ(CuKα) = 0.840 mm−1, 19511 reflections measured, 3394 independent reflections (Rint = 0.0362). The final R1 values were 0.0389 (I > 2σ(I)). The final wR(F2) values were 0.0999 (I > 2σ(I)). The final R1 values were 0.0389 (all data). The final wR(F2) values were 0.0999 (all data). The goodness of fit on F2 was 1.123.
The data were collected using a Bruker APEX DUO diffractometer with Cu Kα radiation. The crystal structures were solved by direct methods using the SHELXS-97 program28 and refined anisotropically by least-squares method using the SHELXL-97 refinement package.28 ESI.†
ECD calculation of compound 3
Conformational analysis of the S-enantiomer was carried out via Monte Carlo searching with the MMFF94 molecular mechanics force field using the Spartan 10 software.29 The lowest-energy conformers having relative energies within 2 kcal mol−1 (ESI, Fig. S32 and Table S8†) were re-optimized using DFT at the B3LYP/6-31+G (d, p) level in vacuum with the Gaussian 09 program.30 The B3LYP/6-31G+(d, p) harmonic vibrational frequencies were further calculated to confirm their stability. The energies, oscillator strengths, and rotational strengths of the electronic excitations were calculated using the TDDFT methodology at the B3LYP/6-311++G (2d, 2p) level in vacuum. Then, the ECD spectra were simulated by the Gausssum 2.25 program31 (σ = 0.3 eV). The final ECD spectra of (S)-3 was obtained according to the Boltzmann distribution theory and their relative Gibbs free energy (ΔG).Hepatoprotective activity assay
Cytotoxic activity assay
The cytotoxic effects were determined by a MTT colorimetric assay in human hepatocellular carcinoma cell line HePG-2, lung cancer cell lines A549 and NCI-H1975, breast cancer cell lines MDA-MB-231 and MCF-7, and ovarian cancer cell line Hey. Each cell suspension of 1 × 104 cells per mL in DMEM containing 10% fetal bovine serum (v/v) was seeded in 96-well plates and cultured for 24 h in 5% CO2 at 37 °C. After fresh medium (100 μL) containing test sample was added, the cells were cultured for 72 h. Then, the medium was changed into a fresh one containing 5 mg mL−1 MTT. After 4 h incubation at 37 °C, the supernatant was removed and 150 μL of DMSO was added to dissolve formazan crystals. The optical density (OD) value of the formazan solution was measured on a microplate reader at 490 nm. Each assay was replicated three times. The effect of the compounds on tumor cells viability was calculated and expressed by IC50 of each cell line.Conclusions
Although lignan molecule is small and contains only two C6–C3 units, many subtypes of classical lignans, neolignans, and oxyneolignans have been found in stems, bark, leaves, seeds, fruits, roots, and rhizomes of plants. In this study, two other subtypes of neolignans were obtained from a hepatoprotective water decoction of P. chinense. We suggested that the unprecedented neolignane skeletons were likely derived from 8,3′-neolignanes. Significantly, the co-occurrence of these unusual neolignans (1–4) in the genus Penthorum, whose taxonomy is still in dispute, may constitute evidence that Penthorum should be considered a distinct family in the APG system. In addition, only compound 1 displayed hepatoprotective activity against acetaminophen-induced damage and selective cytotoxicity against Hey cancer cell. These findings prompt us to pay more attentions to these unusual types of neolignans in Penthorum and their structure–activity relationships in future.Acknowledgements
Financial support from the Sichuan Science and Technology Support Program (Grant No. 2014SZ0134) is acknowledged.Notes and references
- M. Saleem, H. J. Kim, M. S. Ali and Y. S. Lee, Nat. Prod. Rep., 2005, 22, 696–716 RSC.
- J. Y. Pan, S. L. Chen, M. H. Yang, J. Wu, J. Sinkkonen and K. Zou, Nat. Prod. Rep., 2009, 26, 1251–1292 RSC.
- J. G. Shi, Chemistry of Lignans, Chemical Industry Press, Beijing, 2010, pp. 223–325 Search PubMed.
- G. P. Moss, Pure Appl. Chem., 2000, 72, 1493–1523 CrossRef CAS.
- M. Wang, X. Wu, Y. Jiang and D. Y. Zhang, Food Drug, 2013, 15, 202–205 CAS.
- Q. Lu, M. H. Jiang, J. G. Jiang, R. F. Zhang and M. W. Zhang, J. Agric. Food Chem., 2012, 60, 11097–11103 CrossRef CAS.
- Y. C. He, Y. Zou, C. Peng, J. L. Liu, C. J. He, L. Guo, X. F. Xie and L. Xiong, Fitoterapia, 2015, 100, 7–10 CrossRef CAS.
- T. Zhang, Y. M. Chen and G. L. Zhang, J. Integr. Plant Biol., 2007, 49, 1611–1614 CrossRef CAS.
- F. Zhang, J. S. Wang, Y. C. Gu and L. Y. Kong, J. Nat. Prod., 2012, 75, 538–546 CrossRef CAS PubMed.
- J. K. Woo, C. K. Kim, S. H. Kim, H. Kim, D. C. Oh, K. B. Oh and J. Shin, Org. Lett., 2014, 16, 2826–2829 CrossRef CAS PubMed.
- M. P. Yuldashev, Chem. Nat. Compd., 1998, 34, 508–509 CrossRef CAS.
- Y. Kong, Y. J. Fu, Y. G. Zu, F. R. Chang, Y. H. Chen, X. L. Liu, J. Stelten and H. M. Schiebel, Food Chem., 2010, 121, 1150–1155 CrossRef CAS PubMed.
- C. W. Li and C. B. Cui, Molecules, 2014, 19, 21363–21377 CrossRef CAS.
- M. Wang, Y. Jiang, H. L. Liu, X. Q. Chen, X. Wu and D. Y. Zhang, Nat. Prod. Res., 2014, 28, 70–73 CrossRef CAS PubMed.
- C. H. Ng, K. Rullah, M. F. F. M. Aluwi, F. Abas, K. W. Lam, I. S. Ismail, R. Narayanaswamy, F. Jamaludin and K. Shaari, Molecules, 2014, 19, 11645–11659 CrossRef PubMed.
- X. D. Cao, Z. S. Ding, F. S. Jiang, X. H. Ding, J. Z. Chen, S. H. Chen and G. Y. Lv, Nat. Prod. Res., 2012, 26, 2089–2094 CrossRef CAS PubMed.
- M. O. M. Marques, M. C. C. P. Gomes, M. Yoshida and O. R. Gottlieb, Phytochemistry, 1992, 31, 275–277 CrossRef CAS.
- V. S. Prakash Chaturvedula, S. M. Hecht, Z. Gao, S. H. Jones, X. Feng and D. G. I. Kingston, J. Nat. Prod., 2004, 67, 964–967 CrossRef CAS.
- S. X. Zhang, K. Chen, X. J. Liu, D. C. Zhang, T. W. Tao-Wiedmann, S. L. Leu, A. T. McPhail and K. H. Lee, J. Nat. Prod., 1995, 58, 540–547 CrossRef CAS.
- F. P. Lee, Y. C. Chen, J. J. Chen, I. L. Tsai and I. S. Chen, Helv. Chim. Acta, 2004, 87, 463–468 CrossRef CAS PubMed.
- M. Kuroyanagi, K. Yoshida, A. Yamamoto and M. Miwa, Chem. Pharm. Bull., 2000, 48, 832–837 CrossRef CAS.
- Y. M. Chi, F. Hashimoto, W. M. Yan and T. Nohara, Phytochemistry, 1997, 46, 763–769 CrossRef CAS.
- T. Iida and K. Ito, Phytochemistry, 1983, 22, 763–766 CrossRef CAS.
- L. M. X. Lopes, M. Yoshida and O. R. Gottlieb, Phytochemistry, 1984, 23, 2021–2024 CrossRef CAS.
- R. Takeda, J. Hasegawa and M. Shinozaki, Tetrahedron Lett., 1990, 31, 4159–4162 CrossRef CAS.
- H. Morita, E. Kishi, K. Takeya and H. Itokawa, Phytochemistry, 1992, 31, 3993–3995 CrossRef CAS.
- B. N. Su, Y. Takaishi and T. Kusumi, Tetrahedron, 1999, 55, 14571–14586 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- Spartan 10, Wavefunction, Inc., Irvine, CA Search PubMed.
- Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford CT, 2010, A full list of authors can be found in the ESI Search PubMed.
- Gausssum 2.25: N. M. O'Boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29, 839–845 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1D, 2D NMR, and HRESIMS IR spectra for compounds 1–4, X-ray data for 2, and ECD calculation details for 3. CCDC 1414938. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra15982b |
| This journal is © The Royal Society of Chemistry 2015 |