
Robust poly(lactic acid) membranes improved by polysulfone-g-poly(lactic acid) copolymers for hemodialysis†
Xuemin Yua,
Fu Liu*a,
Linghui Wangb,
Zhu Xionga and
Yunze Wanga
aNingbo Institute of Materials Technology & Engineering, Chinese Academy of Sciences, Ningbo, 315201, P. R. China. E-mail: fu.liu@nimte.ac.cn; Tel: +86-574-86685256
bSchool of Chemical Engineering, Ningbo University of Technology, Ningbo, 315016, P. R. China
First published on 10th September 2015
Abstract
Poly(lactic acid) (PLA) is a sustainable membrane candidate for liquid separation and purification. However, the inherent brittleness restrains its practical application, especially for a porous membrane with thin thickness and high porosity. We aim to prepare PLA membranes with controlled pore structure and dialysis performance with improved mechanical and thermal stability by incorporating polysulfone-graft-poly(lactic acid) (PSf-g-PLA) copolymer. Different from the common rubbery elastomer, the brush-like PSf-g-PLA copolymer with a rigid backbone chain and soft side chains was elaborately synthesized to toughen and modify PLA membranes via a phase inversion process. 1H NMR, FTIR and GPC were conducted to determine the structure and molecular weight of the PSf-g-PLA. The influences of chloromethylation substitution and the content of copolymer on the membrane microstructure, the mechanical and thermal stability, and the dialysis performance were investigated in detail. It was demonstrated that the modified PLA membrane exhibited a pure water flux of 54 L m−2 h−1, 95% rejection to BSA, and 65% and 18% clearance of urea and lysozyme, respectively. Besides, both mechanical and thermal stability of the modified PLA membrane were improved by incorporating brush-like PSf-g-PLA.
1. Introduction
Poly(lactic acid) (PLA) is a bio-based thermoplastic aliphatic polyester originating from renewable resources.1,2 As a carbon neutral, sustainable and environmentally friendly material, PLA is promising to replace or replenish petroleum-based plastics in some applications such as fibers, packaging, films and membranes etc.3–7 However, the inherent brittleness and low glass transition temperature of PLA impede its wide applications, especially for a porous membrane with high porosity and thin thickness.8–10 Thereafter, how to improve the resistance to heat, stretching and sterilization is a great challenge in order to achieve the practical application of PLA membranes.Previous research on PLA membranes are mainly focused on the porous structure control and related filtration performances. Tanaka et al. have succeeded in the preparation of PLA depth filter microfiltration membranes serving as bacterial retaining and screen filters via thermally induced phase separation with drying11 or a process that combined non-solvent induced and thermally induced phase separation.12 PLA hollow fiber membranes have also been prepared by phase separation method with DMSO as bore fluid and polyethylene glycol as additives, which exhibited high water permeability and good separation performance.13 Besides, Bettahalli developed PLA hollow fiber with thin dense top-layer and spongy sub-layer for artificial vasculature in tissue engineering scaffolds.14 In our previous work, we explored the possibility of producing PLA porous membranes via phase inversion for hemodialysis. We accomplished the controllable transition from finger-like pores to interconnected pores of PLA membranes via phase inversion by tuning the molecule weight of poly(ethylene oxide) (PEO), the coagulation intensity, and the mass fraction of PEO to PLLA.15 Furthermore, heparin was immobilized to PLA membrane surface through dopamine bridge to improve the compatibility. The in vitro results demonstrated that surface heparinization improved the hemocompatibility of PLA membrane, suppressed the adhesion of platelet, extended plasma recalcification time, and also decreased hemolysis ratio.16 The surface zwitterionization was achieved by dopamine inspired bromoalkyl initiator anchoring and subsequent atom transfer radical polymerization (ATRP) of zwitterionic poly(sulfobetaine methacrylate) (PSBMA).17 The modified PLA membrane exhibited good dialysis performances. When the polymerization time of PSBMA was 1 h, the water flux was 184 L (m−2 h−1), while BSA retention reached up to 90%, and the clearance of urea and creatinine was maintained at 66% and 60%, respectively.
As we all know, extensive efforts have been devoted to modifying PLA composites. Rubbery elastomers with Tg lower than room temperature are mostly applied to toughen PLA composites. CPU,18 POE,19 PCL,20,21 PBAT,22–24 PPC25 etc. are commonly blended with PLA to improve the toughness by producing silver lines and shearing zone, which consequently absorb the impact energy of PLA composites. In most cases, the elastomers are usually incompatible with PLA, therefore, many investigations have been done to improve the compatibility of elastomers. For example, glycidyl methacrylate grafted POE (mPOE) was used as reactive compatilizers to toughen PLA. The elongation at break of PLA blends increased from 21% to 282% when PLA/mPOE is 55/45, while the tensile strength decreased from 74.5 MPa to 36.3 MPa.19 Besides, P(LA-co-CL),26–28 lysine triisocyanate21 were also used as compatilizers to improve the miscibility between PLA and PCL. Elastomer toughening technique is effective to improve the impact strength and the flexibility of PLA significantly. However, this method is inevitable to reduce the tensile strength due to the incorporation of soft segments of rubbery polymers. The hard segments of PU can overcome the negative effect of elastomer toughening on the stiffness of PLA blends.29,30 Zhang prepared polylactide/bio-based thermoplastic polyurethane/free radical initiator ternary blends. The hard-segment of polyurethane maintained the tensile strength of the cross-linked PLA composites, while improved the elongation at break up to 36 times compared to pristine PLA.29 Cold drawing mechanism is thought to be responsible for the toughening of rigid-particles. Due to the different Young's modulus and Poisson ratio of the dispersed phase and continuous phase (Ed > Ec, νd > νc), a high static pressure is produced in the two-phase interface. On the premise of good interfacial adhesion between matrix and dispersed particles, the stress is concentrated on the dispersed particles, leading to the toughness improvement of PLA blend.
According to the critical review on PLA membranes and composite, it is surprising that the mechanical and thermal stability issue of PLA membrane has actually been ignored. Although versatile studies have been conducted to toughen PLA composites, toughening mechanism and performances would be totally different in case of porous membranes with micro-/nano-microstructure and thin thickness (e.g. 40–200 μm). In fact, improving the mechanical and thermal resistance has been a critical issue to break through the bottleneck of PLA membranes, as the porous membrane with thin thickness has higher requirement for the mechanical and thermal stability during the hollow fiber spinning, stretching, drying, sterilization, storage and operation. To the best of our knowledge, we first synthesized brush-like copolymer PSf-g-PLA with rigid backbone chain and flexible side chains to improve the mechanical and thermal stability of PLA membrane via phase inversion. The influence of chloromethylation substitution and the content of copolymer on the membrane microstructure, the mechanical properties, the thermal resistance, water flux and selective separation to BSA, urea and lysozyme was investigated in details.
2. Experimental section
2.1 Materials
Poly(lactic acid) (PLA) (Mn = 110 K–120 K Dalton, Natural Works, US) dried at 80 °C for 24 h was used to prepare membrane. Polysulfone (PSf) (Udel P3500 LCD MB7) was provided by Solvay Advanced Polymers. Paraformaldehyde, tin(IV) chloride (SnCl4), chlorotrimethylsilane ((CH3)3SiCl), bovine serum albumin (BSA, Mw 67![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), urea and lysozyme were purchased from Aladdin. Dimethylsulfoxide dehydrated (DMSO), chloroform (CHCl3), ethylenediamine (EDA), methanol and ethanol obtained from Sinopharm Chemical reagent Co. were all used as received.
000), urea and lysozyme were purchased from Aladdin. Dimethylsulfoxide dehydrated (DMSO), chloroform (CHCl3), ethylenediamine (EDA), methanol and ethanol obtained from Sinopharm Chemical reagent Co. were all used as received.
2.2 Synthesis and characterization of PSf-g-PLA
Chloromethylation of PSf (CMPSf) was performed according to a conventional procedure as follows:31 50 g of PSf was first dissolved into 750 mL of chloroform in a stirring round bottom flask equipped with a reflux condenser, 10 g of paraformaldehyde, 0.875 g of anhydrous SnCl4, 50 mL of (CH3)3SiCl were added. The reaction was conducted at 85 °C for 8 h, 12 h, 26 h and 34 h, respectively to produce CMPSf with different percent of chloromethylation per repeat unit of polysulfone (DS, degree of substitution). The resultant solution after desired reaction time was poured into methanol and the polymer was precipitated and filtered. Subsequently, the polymer was redissolved in chloroform and precipitated in methanol to be further purified, then filtered and dried under vacuum for 24 h. The resultant sample was named as CMPSf-08, CMPSf-12, CMPSf-26 and CMPSf-34 respectively, in correspondence with the reaction time.Amino functionalized polysulfone (PSf-EDA) was conducted by dissolving 10 g of CMPSf and 10 g of EDA into 100 g of DMAc. The –CH2Cl in CMPSf can react with EDA at room temperature. After 4 h, the mixture was precipitated in water. After filtration, the polymer was re-dissolved in DMAc and precipitated in water, then filtered and dried under vacuum for 24 h. The PSf-EDA was named as PSf-EDA-08, PSf-EDA-12, PSf-EDA-26 and PSf-EDA-34, respectively.
The synthesis of PSf-g-PLA was based on the aminolysis reaction of PLA with the amino functionalized polysulfone (PSf-EDA). For example, PSf-EDA-26 was added to a solution of PLA in DMSO. The reaction was proceeded under stirring at 80 °C for 5 h. The resultant solution was ready for membrane casting after being kept still to remove air bubbles. The total weight of PSf-EDA-26 and PLA was 18 g, and the weight of DMSO was 82 g. The composition of the casting solution containing PSf-EDA, PLA and DMSO are listed in Table 1.
| PSf | WPSf-EDA/g | WPLA/g | WDMSO/g | W* (g/g) |
|---|---|---|---|---|
| a W*: the weight ratio of PSf-EDA to the weight of PSf-EDA and PLA. | ||||
| 0 | 18.00 | 82.00 | 0% | |
| Pure PSf | 0.90 | 17.10 | 82.00 | 5% |
| PSf-EDA-08 | 0.90 | 17.10 | 82.00 | 5% |
| PSf-EDA-12 | 0.90 | 17.10 | 82.00 | 5% |
| PSf-EDA-26 | 0.90 | 17.10 | 82.00 | 5% |
| PSf-EDA-34 | 0.90 | 17.10 | 82.00 | 5% |
| PSf-EDA-26 | 0.18 | 17.82 | 82.00 | 1% |
| PSf-EDA-26 | 0.54 | 17.46 | 82.00 | 3% |
| PSf-EDA-26 | 1.80 | 16.20 | 82.00 | 10% |
CMPSf and PSf-EDA were characterized by 1H NMR spectra, recorded on AVANCE III/400 MHz NMR spectrometer at room temperature in CDCl3 with Si(CH3)4 as an internal standard. The peak at 4.53 ppm represented the –CH2Cl protons (donated as (a)), while the peak at 8.0 ppm assigned to the four meta protons on the phenyl ring closest to the sulfonyl group (denoted as (b)). The percent of chloromethylation per repeat unit (DS, degree of substitution) can be calculated by the proportion of the peak areas at 4.53 ppm (A) and 7.86 ppm (B) as shown in eqn (1):
| DS = 2A/B | (1) |
PSf, PLA resin and PSf-g-PLA after purification were measured by FTIR to determine the chemistry structure. FTIR spectra were recorded from 400 to 4000 cm−1 with Fourier transform infrared spectra (Thermo-Nicolet 6700, US). The molecular weight of PSf, PLA, CMPSf, PSf-EDA and PSf-g-PLA were measured by GPC. The GPC experiments were carried out by a Waters 1515 GPC system eluted at 1.0 mL·min−1 with THF at 40 °C. The system was calibrated by narrow polystyrene standards.
2.3 Preparation of PLA membranes
PLA membranes were prepared via a typical phase inversion procedure as follows: the prepared solution containing PSf-EDA, PLA and DMSO was directly casted onto a glass plate by a blade with a thickness of 200 μm, which was immediately immersed into a water bath at 30 °C for 10 min for coagulation. After the total solidification, the membranes were soaked into deionized water to remove the residue solvent for further characterization. The detailed recipe of casting solution containing PSf-EDA, PLA and DMSO is listed in Table 1.2.4 Characterization of PLA membranes
The mechanical properties were characterized on Instron 5567 instrument using sample films (10 × 50 mm2) under a stretching rate of 10 mm min−1. The morphology of the cross section and top surface of the membranes were observed by a scanning electron microscope (SEM, Hitachi S-4800, Japan). Samples of the cross section were quenched by liquid nitrogenous gas. All membrane samples were attached to the sample supports and sputtered with gold for 2 min. Surface roughness was measured by atomic force microscopy (AFM, Dimension 3100 V, Veeco, US).The thermal resistance of the porous membrane was characterized by putting membrane samples (10 × 100 mm2) in an oven at 50 °C, 60 °C and 70 °C for 2 h and then measuring the length remain.
The shrinkage was calculated as eqn (2):
 | (2) |
The pure water flux was measured by using a filtration apparatus (Saifei Company, China) with an effective membrane area of 24 cm2. The membranes were pre-pressured at 0.2 MPa about 30 min by deionized water, then the stable flux was recorded every 5 min at 0.1 MPa.
The simulated dialysis performance of the membranes was measured at 37 °C using a mixed solution of urea (1.5 g L−1), lysozyme (0.04 g L−1) and BSA (1.0 g L−1) in physiological saline (0.9 wt%) as simulative blood (flow rate 100 mL min−1). Distilled water played the role of simulative dialysate (flow rate 300 mL min−1). The effective membrane area was 64 cm2. After dialyzing for 6 h, the test solution was taken out from the simulative blood.17 The concentration change of urea was evaluated by mixing 1.5 mL simulative blood with 0.8 mL PDAB solution (0.1 M, dissolved in 1.2 M H2SO4) and 1.7 mL deionized water, then the urea concentration was detected at 410 nm by the UV-vis spectrophotometer Lambda 950 (Perkin-Elmer, America). Lysozyme and BSA were measured at 278 nm and 280 nm by a UV-vis spectrophotometer Lambda 950 (Perkin-Elmer, America). The urea clearance (Clearanceurea) was calculated as eqn (3):
 | (3) |
The lysozyme clearance (Clearancelysozyme) was calculated as eqn (4):
 | (4) |
The BSA retention (RetentionBSA) was calculated by eqn (5):
 | (5) |
3. Results and discussion
3.1 Chemistry of copolymer PSf-g-PLA
The schematic diagram of synthesis of PSf-g-PLA was shown in Fig. 1. PSf was first activated by chloromethylation, and then the activated CMPSf reacted with excessive ethylenediamines (EDA) to produce amino functionalized polysulfone (PSf-EDA). The primary amine bonded to PSf-EDA attacked the carbonyl carbon in the PLA through aminolysis reaction to produce the copolymer with polysulfone as the backbone chain and PLA as the side chain. | ||
| Fig. 1 The synthetic mechanism of PSf-g-PLA via aminolysis. | ||
A representative 1H NMR spectrums of CMPSf was shown in Fig. 2. The peak at 4.53 ppm represented the –CH2Cl protons (donated as (a)), while the peak at 8.0 ppm arised from the four meta protons on the phenyl ring closest to the sulfonyl group (denoted as (b)). The peak (a) intensity increased with the reaction time, and the DS value, calculated by eqn (1), was 8%, 15%, 24% and 48%, for CMPSf-08, CMPSf-12, CMPSf-26 and CMPSf-34, respectively. Besides, the 1H NMR spectrum of the PSf-EDA-26 showed that the peak at 4.53 ppm was distinctly shifted to 3.66 ppm, representing the –CH2NH– donated as (c). Furthermore, the new peaks emerged at 2.56 ppm and 2.69 ppm represented the –CH2CH2NH2 donated as (d) and (e). Therefore, all above results certainly confirmed that the –Cl of CMPSf was completely substituted by –NHCH2CH2NH2 after reaction between CMPSf and EDA.
 | ||
| Fig. 2 1H NMR spectrums of the CMPSf and PSf-EDA. | ||
The chemistry of copolymer PSf-g-PLA, PSf and PLA was characterized by FTIR as shown in Fig. 3(A), similar to the FTIR spectrums of original PSf and PLA, PSf-g-PLA showed absorption band at 1759 cm−1 ascribed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of PLA, 1582 cm−1 ascribed to CC of PSf, 1323 cm−1 ascribed to SO2 asymmetric stretch of PSf, 1147 and 1170 cm−1 ascribed to SO2 symmetric stretch of PSf, which indicated that the copolymer was composed of PLA and PSf. Besides, the newly appeared absorption band at 3095 cm−1 and 1680 cm−1 was attributed to the functional linking amide group of –NH–CO– between PSf and PLA, which confirmed the formation of brush-like copolymer PSf-g-PLA. The primary amine (–NH2) bonded to PSf-EDA attacked the carbonyl carbon in the PLA through nucleophilic substitution to produce requisite copolymer PSf-g-PLA. It should be noted that the cleavage of ester bonds would induce the degradation of PLA inevitably to some extents due to the intrinsic mechanism of aminolysis.32,33
O of PLA, 1582 cm−1 ascribed to CC of PSf, 1323 cm−1 ascribed to SO2 asymmetric stretch of PSf, 1147 and 1170 cm−1 ascribed to SO2 symmetric stretch of PSf, which indicated that the copolymer was composed of PLA and PSf. Besides, the newly appeared absorption band at 3095 cm−1 and 1680 cm−1 was attributed to the functional linking amide group of –NH–CO– between PSf and PLA, which confirmed the formation of brush-like copolymer PSf-g-PLA. The primary amine (–NH2) bonded to PSf-EDA attacked the carbonyl carbon in the PLA through nucleophilic substitution to produce requisite copolymer PSf-g-PLA. It should be noted that the cleavage of ester bonds would induce the degradation of PLA inevitably to some extents due to the intrinsic mechanism of aminolysis.32,33
 | ||
| Fig. 3 (A) FTIR spectrums of PSf, PLA and PSf-g-PLA and (B) normalized GPC curves for PSf, PLA, CMPSf-26, PSf-EDA-26, and the modified PLA membrane. | ||
Furthermore, the molecular weight of PSf, PLA, CMPSf, PSf-EDA and PSf-g-PLA was evaluated by GPC as shown in Fig. 3(B). PSf, CMPSf-26 and PSf-EDA-26 all showed an unimodal GPC trace, in addition, the molecular weight of CMPSf-26 and PSf-EDA-26 increased slightly compared to PSf due to the possible crosslinking between PSf chains,34 moreover, the modified PLA membrane showed a bimodal distribution. The higher peak with shorter elution time indicated PSf-g-PLA with higher molecular weight was produced by grafting PLA to PSf, while the lower peak with longer elution time indicated the presence of PLA. Therefore, modified PLA membrane was composed of PSf-g-PLA and PLA. The composition of the modified PLA membrane containing PLA and PSf-g-PLA, listed in Table 2, was calculated by DS value and content of PSf-EDA as eqn (6) and (7), and the detailed calculation could be seen in ESI:†
 | (6) |
| WPLA = (1 − WPSf-g-PLA) × 100% | (7) |
| PSf-EDA | DS value [%] | WPSf-EDA [wt%] | WPSf-g-PLA [wt%] | WPLA [wt%] |
|---|---|---|---|---|
| PSf-EDA-08 | 8 | 5 | 51.5 | 48.5 |
| PSf-EDA-12 | 15 | 5 | 66.0 | 34.0 |
| PSf-EDA-26 | 24 | 5 | 75.5 | 24.5 |
| PSf-EDA-34 | 48 | 5 | 85.9 | 14.1 |
| PSf-EDA-26 | 24 | 1 | 36.2 | 63.8 |
| PSf-EDA-26 | 24 | 3 | 63.9 | 36.1 |
| PSf-EDA-26 | 24 | 10 | 87.3 | 12.7 |
3.2 Morphology of modified PLA membranes
The disperse morphology of copolymer in the matrix and the interfacial compatibility with PLA bulk influenced the membrane stability and filtration performances substantially. Obviously, the chloromethylation of PSf influenced the subsequent amination of PSf and the resultant morphology. The cross section morphology of modified PLA membrane with different DS value was shown in Fig. 4 (left). The control PLA/PSf membrane without chloromethylation showed a random disperse of PSf particles with the size up to 30 μm caged in the macrovoids. The clear interface between two phases implied the weak interaction and poor compatibility between PSf and PLA. In case of modified PLA membrane with DS value of 8%, the copolymer was still present in the membrane matrix, however, the particle was reduced to ∼1.5 μm and rooted in PLA bulk. Besides, the interfaces between two phases were narrowed to some extents, which suggested the two phase compatibility was significantly improved. With further increasing the DS value to 15%, the copolymer particle was reduced to ∼0.5 μm. When the DS value increased to 24% and 48%, the copolymer displayed a excellent compatibility with PLA bulk and was completely disappeared and unable to be distinguished by SEM with the magnification of ×40 K. | ||
| Fig. 4 SEM images of the cross-section for modified PLA membranes with different DS value (left) and different PSf-EDA-26 content (right). | ||
Furthermore, the disperse of PSf-EDA-26 in the PLA membrane cross section was shown in Fig. 4 (right). When the PSf-EDA-26 content was lower than 5 wt%, the modified membrane showed good compatibility and no dispersed particles or interfaces between two phases were found in the membrane. When the PSf-EDA-26 content was 10 wt%, the copolymer particles were clearly embedded in the membrane bulk due to the incompatibility and aggregation.
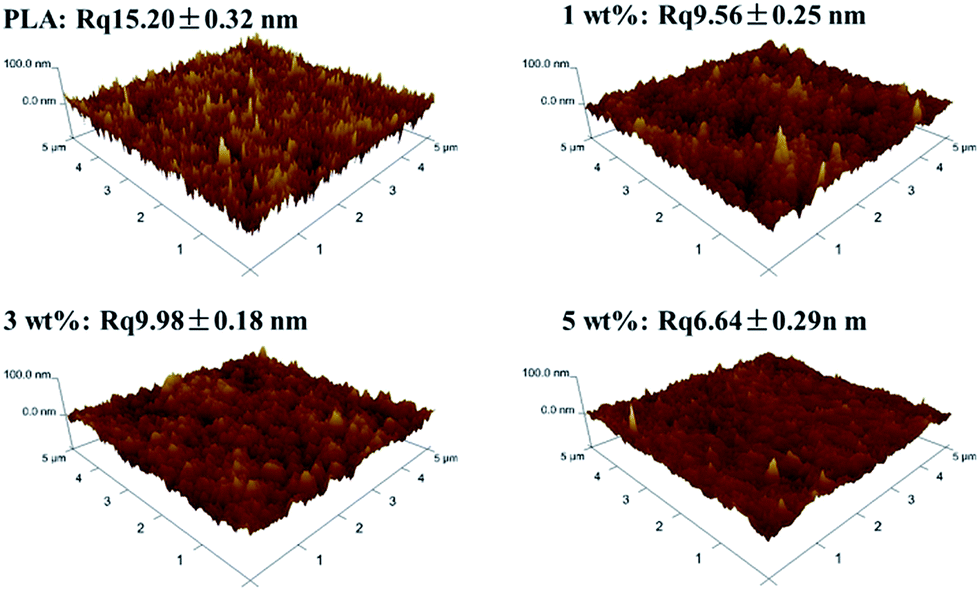
Besides the cross section, the top surface as the selective layer was also influenced by the content of PSf-EDA-26. As shown in Fig. 5, the top surface of PLA membrane was quite porous. The pore sizes were measured by image pro plus of different pores from SEM images. The images of pore size distribution were shown in Fig. S1 in ESI.† The average radium (Ra) of pore in PLA membrane was 22.4 ± 1.5 nm.35 While the pore size of the top surface was smaller with adding 1 wt% PSf-EDA-26 (Ra = 10.2 ± 1.2 nm). With further adding 3 wt% and 5 wt% PSf-EDA-26, the membrane surface was getting denser and Ra was 9.2 ± 0.8 nm and 4.7 ± 0.6 nm, respectively. The roughness reduction of modified PLA membranes also verified the morphology evolution as shown in Fig. 6. The roughness of pristine PLA is 15.2 nm, in comparison, the modified PLA membrane with 5% PSf-EDA-26 exhibited a substantially reduced Rq 6.64 nm, indicating a denser and smoother surface. The morphology variation was closely related with the phase separation. The higher molecular weight copolymer PSf-g-PLA fortified the entanglement with PLA segments and increased the viscosity of the casting solution. It has been generally accepted that higher polymer concentration was obtained at the polymer/bath interface prior to immersion in the coagulation bath. The diffusion of non-solvent into the polymer solution through the interface was slowed down by the higher viscosity of copolymer solution. Therefore, the delayed demixing was thought to dominate the phase separation and less porous membranes were obtained.36–38
 | ||
| Fig. 5 SEM images of the top surface for modified PLA membranes with different PSf-EDA-26 content. | ||
 | ||
| Fig. 6 Surface AFM images of membranes with different PSf-EDA-26 contents. | ||
3.3 Stability of modified PLA membranes
The influence of DS value on the mechanical stability of the modified PLA membranes was illustrated in Fig. 7(A). The content of PSf-EDA was set as 5 wt%. The tensile strength and elongation at break of the control PLA/PSf blend membrane was only 2.34 MPa and 18.2%, respectively. With increasing DS value, the tensile strength continuously increased from 2.47 MPa (8% DS value) to 3.74 MPa (48% DS value) and the elongation at break increased from 21.5% (8% DS value) to 48.0% (24% DS value), which was three times higher than the original PLA/PSf membrane, and then decreased to 41.8% (48% DS value) possibly due to the serious ammonolysis of PLA. As a contrast, the tensile strength of pure PLA membrane was 2.91 MPa, and the elongation at break was only 23.2%. Both tensile strength and elongation at break of PLA membrane modified by brush-like copolymer PSf-g-PLA was improved simultaneously. For example, in case of membrane with the DS value of 24%, the tensile strength and elongation at break reached up to 3.57 MPa and 48.0%. From the evolution of micro-morphology of modified PLA membrane as shown in Fig. 4 (left), it could be inferred that the poor disperse of copolymer with DS value lower than 15% induced more stress concentration sites and reduced the mechanical strength accordingly compared to pure PLA membrane. Whereas, with increasing the chloromethylation, more flexible PLA side chains were grafted onto the rigid backbone chain of PSf, therefore, high flexibility improved the compatibility of copolymer. The excellent compatibility of copolymer with DS value higher than 15% guaranteed the uniform entanglement and better interfacial adhesion between more flexible brush-like copolymer and PLA, which enhanced the strength and flexibility of the modified membranes subsequently.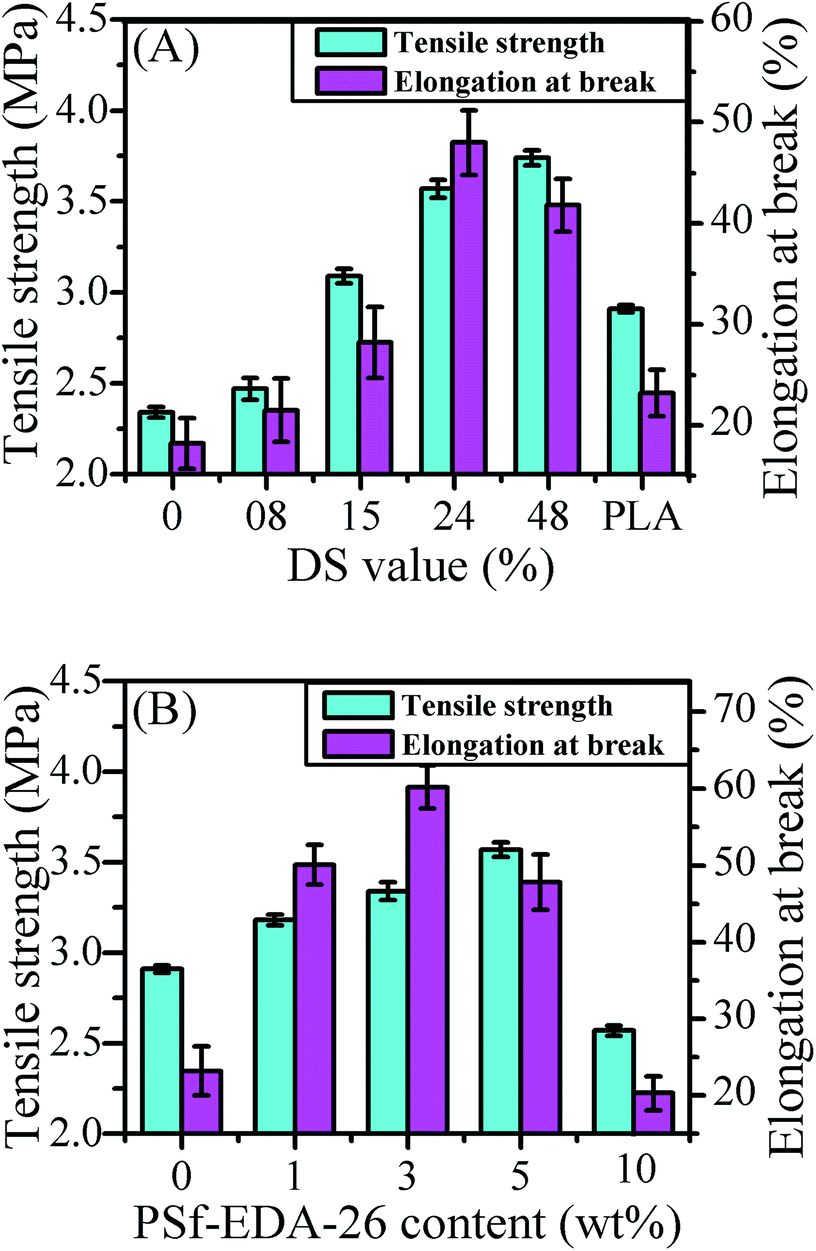 | ||
| Fig. 7 The mechanical properties of modified PLA membrane with different DS value (A) and different PSf-EDA-26 content (B). | ||
The influences of PSf-EDA-26 content with DS value of 15% on membrane properties were investigated. The mechanical stability of membranes varied with different PSf-EDA-26 contents as shown in Fig. 7(B). Firstly, the tensile strength increased from 2.91 MPa (neat PLA) to 3.57 MPa for membrane with 5 wt% PSf-EDA-26 (containing 75.5% PSf-g-PLA), then decreased from 3.57 MPa to 2.57 MPa for membrane with 10 wt% PSf-EDA-26 (containing 87.3% PSf-g-PLA) due to the incompatibility and aggregation of copolymer as shown in Fig. 4 (right). Similarly, the elongation at break increased from 23.2% (neat PLA) to 60.2% for membrane with 3 wt% PSf-EDA-26 (containing 63.9% PSf-g-PLA), and then decreased to 20.3% for membrane with 10 wt% PSf-EDA-26 (containing 87.3% PSf-g-PLA).
The mechanism of toughening PLA membrane by brush-like copolymer PSf-g-PLA was suggested in Fig. 8. Unlike the toughening PLA by rubbery elastomers, the modified PLA membrane was toughened by the brush-like copolymer PSf-g-PLA consist of both hard domain and soft domain, which dominated by rigid polysulfone backbone and flexible PLA chains respectively. When the chloromethylation of PSf was lower than 15% or the content of PSf-EDA-26 was higher than 10 wt%, the copolymer particle had a reduced compatibility with PLA bulk, which was verified by the bigger particle size. The increased interfaces from d to d′ between hard domain and the soft domain also supported the compatibility variation, as suggested in Fig. 8. The hard domain kept its conformation and stiffness under stress to increase the tensile strength. While the stretched soft domain increased the interfaces, which worked as stress concentration points in the PLA membrane to absorb the stretching energy and hinder the crack growth during the process of shear yielding.18,39 The flexible side PLA chains had a good entanglement with free PLA to anchor the copolymer in the membrane matrix and were mainly responsible for the compatibility. Therefore, the elongation at break of the modified PLA membrane was significantly improved without sacrificing the tensile strength with the aid of brush-like copolymer composing of rigid PSf backbone and flexible side PLA chains.
 | ||
| Fig. 8 The mechanisms of toughening the PLA membrane by PSf-g-PLA. | ||
The thermal dimension stability was also crucial to the porous membrane. Especially, there is a urgent requirement for the membrane drying and disinfection at high temperature. The poor thermal resistance and low glass transition temperature (Tg ∼ 55 °C) induced the serious shrinkage of PLA membrane under drying, e.g. the shrink rate of control PLA membrane is 22%, 20% and 5% at 70 °C, 60 °C and 50 °C, respectively as shown in Fig. 9. With increasing the contents of PSf-EDA-26, the shrink rate of the modified PLA membrane decreased significantly, indicating the improved dimension and pore structure stability under heat treatment. For example, the modified PLA membrane with 10 wt% PSf-EDA-26 dried at 60 °C and 50 °C demonstrated a reduced shrink rate of 8% and 0%. The flexible side chain PLA provided the good compatibility and uniform distribution of copolymer in the modified PLA membrane, the PSf backbone chain with the high glass transition temperature (Tg ∼ 190 °C) was responsible for the enhancement of thermal stability. Nevertheless, the shrink rate of the modified PLA membrane was as the same as control PLA membrane, when the temperature was higher than 70 °C due to the porous structure and thin thickness.
 | ||
| Fig. 9 The shrink rate of membranes with different PSf-EDA-26 content at 50 °C, 60 °C and 70 °C respectively. | ||
3.4 Filtration performances of modified PLA membranes
Pure water flux of the modified PLA membranes was shown in Fig. 10(A). The water flux was declined with increasing the content of PSf-EDA-26. The pristine PLA membrane demonstrated a water flux of 318 L m−2 h−1, while the modified PLA membrane exhibited a reduced water flux of 143 L m−2 h−1, 54 L m−2 h−1 and 0 L m−2 h−1 with increasing the content of PSf-EDA-26 to 1 wt%, 3 wt% and 5 wt%, respectively. Besides, the selectivity to urea, lysozyme and BSA was measured by a hemodialysis simulation test and the results were shown in Fig. 10(B). The clearance of urea and lysozyme decreased with increasing the content of PSf-EDA, while the BSA rejection increased. The modified PLA membrane with 3 wt% PSf-EDA exhibited water flux of 54 L m−2 h−1, BSA retention of 95%, clearance of urea and lysozyme of 65% and 18%. The permeability and selectivity of PLA membranes were mainly determined by the membrane micro-structure. The modified membrane showed a denser membrane surface and selective layer as shown in Fig. 5 and 6, which reduced the permeability of water, urea and lysozyme and enhanced the retention of BSA accordingly.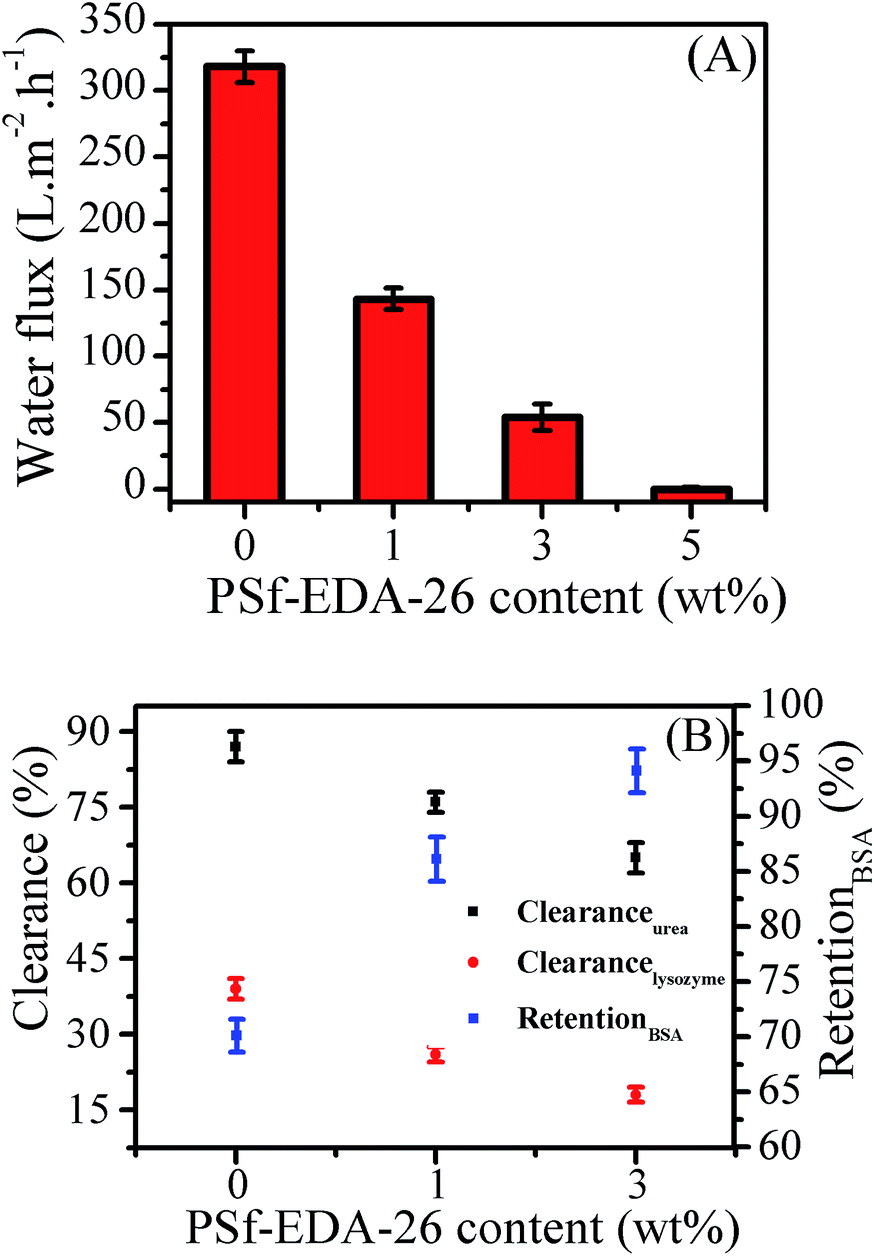 | ||
| Fig. 10 Water flux of membranes (A) and urea clearance (Clearanceurea), lysozyme clearance (Clearancelysozyme) and BSA retention (RetentionBSA) (B) with different PSf-EDA-26 content. | ||
The modified PLA membrane was compared to PSf-based, PES-based and PLA-based membranes reported in literature in terms of mechanical strength, pure water flux and BSA rejection as listed in Table 3. The produced PLA membrane showed an enhanced mechanical strength, especially the elongation at break was 60.2%, which was much higher than the current membranes. Besides, the modified PLA membrane showed a high BSA retention and relatively low permeability, indicating a tight ultrafiltration membrane suitable for hemodialysis. The micro-structure of PLA membrane would be further tuned in later work.
4. Conclusion
The brush-like copolymer PSf-g-PLA was first synthesized to improve the morphology, stability and filtration performance of PLA membrane. Chloromethylation, amination and grafting of PLA was confirmed by the 1H NMR, FTIR and GPC results respectively, which demonstrated that the modified PLA membrane was composed of brush-like copolymer PSf-g-PLA and PLA. With increasing the degree of chloromethylation of PSf, the copolymer exhibited a more uniform disperse and better compatibility with PLA bulk, which enhanced the elongation at break and elongation strength simultaneously. The newly appeared interface between the hard domain and soft domain was thought to be the key mechanism to toughen PLA. The modified PLA membrane showed enhanced elongation at break and tensile strength up to 60.2% and 3.34 MPa. Besides, the modified PLA membrane exhibited a decreased thermal shrinkage. The excellent compatibility between brush-like copolymer PSf-g-PLA an PLA provided a denser and more uniform and smooth surface. The modified PLA membrane exhibited a pure water flux of 54 L m−2 h−1, 95% rejection to BSA, 65% and 18% clearance of urea and lysozyme, respectively, which indicated its potential application as a robust hemodialysis membrane.Acknowledgements
This work is supported by National Natural Science Foundation of China (51473177, 51273211) and Natural Science Foundation of Ningbo (2015A610024).Notes and references
- R. H. Wehrenberg, Mater. Eng., 1980, 94, 63–66 Search PubMed.
- E. Lipinsky and R. Sinclair, Chem. Eng. Prog., 1986, 82, 26–32 CAS.
- R. M. Rasal and D. E. Hirt, Macromol. Biosci., 2009, 9, 989–996 CrossRef CAS PubMed.
- S. S. Ray and M. Okamoto, Macromol. Rapid Commun., 2003, 24, 815–840 CrossRef CAS PubMed.
- Z. Zhang and S.-S. Feng, Biomaterials, 2006, 27, 4025–4033 CrossRef CAS PubMed.
- C. Gottschalk and H. Frey, Macromolecules, 2006, 39, 1719–1723 CrossRef CAS.
- C. Schugens, C. Grandfils, R. Jérôme, P. Teyssie, P. Delree, D. Martin, B. Malgrange and G. Moonen, J. Biomed. Mater. Res., 1995, 29, 1349–1362 CrossRef CAS PubMed.
- N. Zhang, Q. Wang, J. Ren and L. Wang, J. Mater. Sci., 2009, 44, 250–256 CrossRef CAS.
- R. M. Rasal and D. E. Hirt, J. Biomed. Mater. Res., Part A, 2009, 88, 1079–1086 CrossRef PubMed.
- M. Hiljanen-Vainio, P. Varpomaa, J. Seppälä and P. Törmälä, Macromol. Chem. Phys., 1996, 197, 1503–1523 CrossRef CAS PubMed.
- T. Tanaka, M. Ueno, Y. Watanabe, T. Kouya, M. Taniguchi and D. R. Lloyd, J. Chem. Eng. Jpn., 2011, 44, 467–475 CrossRef CAS.
- T. Tanaka, T. Nishimoto, K. Tsukamoto, M. Yoshida, T. Kouya, M. Taniguchi and D. R. Lloyd, J. Membr. Sci., 2012, 396, 101–109 CrossRef CAS PubMed.
- A. Moriya, T. Maruyama, Y. Ohmukai, T. Sotani and H. Matsuyama, J. Membr. Sci., 2009, 342, 307–312 CrossRef CAS PubMed.
- N. Bettahalli, H. Steg, M. Wessling and D. Stamatialis, J. Membr. Sci., 2011, 371, 117–126 CrossRef CAS PubMed.
- A. Gao, F. Liu, H. Shi and L. Xue, J. Membr. Sci., 2015, 478, 96–104 CrossRef CAS PubMed.
- A. Gao, F. Liu and L. Xue, J. Membr. Sci., 2014, 452, 390–399 CrossRef CAS PubMed.
- L.-J. Zhu, F. Liu, X.-M. Yu, A.-L. Gao and L.-X. Xue, J. Membr. Sci., 2015, 475, 469–479 CrossRef CAS PubMed.
- G.-C. Liu, Y.-S. He, J.-B. Zeng, Y. Xu and Y.-Z. Wang, Polym. Chem., 2014, 5, 2530–2539 RSC.
- Z. Su, Q. Li, Y. Liu, G.-H. Hu and C. Wu, Eur. Polym. J., 2009, 45, 2428–2433 CrossRef CAS PubMed.
- V. Vilay, M. Mariatti, Z. Ahmad, K. Pasomsouk and M. Todo, J. Appl. Polym. Sci., 2009, 114, 1784–1792 CrossRef CAS PubMed.
- T. Takayama and M. Todo, J. Mater. Sci., 2006, 41, 4989–4992 CrossRef CAS.
- F. Signori, M.-B. Coltelli and S. Bronco, Polym. Degrad. Stab., 2009, 94, 74–82 CrossRef CAS PubMed.
- R. Al-Itry, K. Lamnawar and A. Maazouz, Polym. Degrad. Stab., 2012, 97, 1898–1914 CrossRef CAS PubMed.
- L. Jiang, M. P. Wolcott and J. Zhang, Biomacromolecules, 2006, 7, 199–207 CrossRef CAS PubMed.
- X. Ma, J. Yu and N. Wang, J. Polym. Sci., Part B: Polym. Phys., 2006, 44, 94–101 CrossRef CAS PubMed.
- S. Aslan, L. Calandrelli, P. Laurienzo, M. Malinconico and C. Migliaresi, J. Mater. Sci., 2000, 35, 1615–1622 CrossRef CAS.
- G. Maglio, A. Migliozzi, R. Palumbo, B. Immirzi and M. G. Volpe, Macromol. Rapid Commun., 1999, 20, 236–238 CrossRef CAS.
- C. H. Kim, K. Y. Cho, E. J. Choi and J. K. Park, J. Appl. Polym. Sci., 2000, 77, 226–231 CrossRef CAS.
- L. Zhang, Z. Xiong, S. S. Shams, R. Yu, J. Huang, R. Zhang and J. Zhu, Polymer, 2015, 64, 69–75 CrossRef CAS PubMed.
- H. Liu and J. Zhang, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 1051–1083 CrossRef CAS PubMed.
- J. Y. Park, M. H. Acar, A. Akthakul, W. Kuhlman and A. M. Mayes, Biomaterials, 2006, 27, 856–865 CrossRef CAS PubMed.
- Y. Zhu, Z. Mao and C. Gao, RSC Adv., 2013, 3, 2509–2519 RSC.
- Y. Hong, C. Gao, Y. Xie, Y. Gong and J. Shen, Biomaterials, 2005, 26, 6305–6313 CrossRef CAS PubMed.
- J. Meng, J. Yuan, Y. Kang, Y. Zhang and Q. Du, J. Colloid Interface Sci., 2012, 368, 197–207 CrossRef CAS PubMed.
- N. Singh, S. M. Husson, B. Zdyrko and I. Luzinov, J. Membr. Sci., 2005, 262, 81–90 CrossRef CAS PubMed.
- P. Vandezande, X. Li, L. E. Gevers and I. F. Vankelecom, J. Membr. Sci., 2009, 330, 307–318 CrossRef CAS PubMed.
- Y. H. See-Toh, F. C. Ferreira and A. G. Livingston, J. Membr. Sci., 2007, 299, 236–250 CrossRef CAS PubMed.
- J. Ren, Z. Li and F.-S. Wong, J. Membr. Sci., 2004, 241, 305–314 CrossRef CAS PubMed.
- J. B. Lee, Y. K. Lee, G. D. Choi, S. W. Na, T. S. Park and W. N. Kim, Polym. Degrad. Stab., 2011, 96, 553–560 CrossRef CAS PubMed.
- Y. Ma, F. Shi, J. Ma, M. Wu, J. Zhang and C. Gao, Desalination, 2011, 272, 51–58 CrossRef CAS PubMed.
- X. J. Huang, D. Guduru, Z. K. Xu, J. Vienken and T. Groth, Macromol. Biosci., 2011, 11, 131–140 CrossRef CAS PubMed.
- Y. Ma, F. Shi, Z. Wang, M. Wu, J. Ma and C. Gao, Desalination, 2012, 286, 131–137 CrossRef CAS PubMed.
- L. Wang, Y. Cai, Y. Jing, B. Zhu, L. Zhu and Y. Xu, J. Colloid Interface Sci., 2014, 422, 38–44 CrossRef CAS PubMed.
- M. Irfan, A. Idris, N. M. Yusof, N. F. M. Khairuddin and H. Akhmal, J. Membr. Sci., 2014, 467, 73–84 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra15816h |
| This journal is © The Royal Society of Chemistry 2015 |