DOI:
10.1039/C5RA15789G
(Paper)
RSC Adv., 2015,
5, 81313-81323
Dihydroisoquinoline copper(II) complexes: crystal structures, cytotoxicity, and action mechanism†
Received
6th August 2015
, Accepted 8th September 2015
First published on 8th September 2015
Abstract
Three new copper(II) complexes of 4,5-methylenedioxy-1-pyridinedihydroisoquinoline (MPDQ), [Cu2(MPDQ)2Cl4] (1), [Cu(MPDQ)(H2O)(SO4)] (2), and [Cu2(MPDQ)2(C2O4)(ClO4)2] (3) were synthesized and characterized. They exhibit enhanced cytotoxicity against the tested human tumor cells BEL-7404, SK-OV-3, HepG2, A549, A375, MGC-803 and NCI-H460 compared to MPDQ and the corresponding copper(II) salts with IC50 values of 1.41–34.54 μM. Complex 1 can induce apoptotic death in BEL-7404 by S cell cycle arrest and complex 1-induced apoptosis, which were involved in an intrinsic pathway, including up-regulating P53 and down-regulating Bcl-2 and mitochondrial membrane potential, leading to sequential activation of caspase-9 and caspase-3. ICP-MS testing implied that the copper(II) complexes could enter cells and DNA was one important target; DNA binding studies revealed that intercalation might be the most probable binding mode of the new Cu(II) complexes with ct-DNA.
Introduction
Cisplatin is a widely used and well-known metal-based drug for cancer therapy but it causes severe side effects and therefore a low administration dosage has to be used.1–3 Therefore, increasing efforts are being made to replace this drug with suitable alternatives, and numerous non-platinum-based antitumor complexes have been synthesized and screened for their anticancer activities.4–9 In addition to Ru(II) complexes, Cu(II) complexes are regarded as the best alternatives to replace cisplatin as anticancer agents.9–11 Copper is known to play a significant role in biological systems and it also has been used as a pharmacological agent.12–14 Synthetic copper(II) complexes have been reported to act as potential anticancer agents, and a number of copper complexes have been found to be active in vivo.15–17
Isoquinolines, a type of alkaloids, are widespread in nature and they exhibit a variety of biological activities, including inhibition of cellular proliferation and cancer development.18–21 For example, berberrubine, a protoberberine alkaloid, showed antitumor activity in animal models22 and 6-alkyl-12-formyl-5,6-dihydroindolo-[2,1-a]-isoquinolines have also been shown to inhibit the growth of human mammary cells.23,24 Since the broad biological effects of isoquinolines have been researched during the past two decades, a series of isoquinoline and related platinum(II) complexes with potent anticancer activity have been reported by Steglich and Farrell et al.25–28 Previous studies from our group revealed that transition metal complexes of natural isoquinoline alkaloids, including the active ingredients liriodenine and oxoglaucine in traditional Chinese medicines, also exhibited promising anticancer activity.7,29–31 3,4-Dihydroisoquinoline (thalpetaline) was isolated from an EtOH-extract of the Chinese medicinal herb T. petaloideum. This compound is known for its efficacy against diarrhea, hepatitis, skin infections and cancer32 and is widely applied in the design of new antitumor agents.33–35 Recently, we utilized 3,4-dihydroisoquinoline as a leading compound in the design and synthesis of one isoquinoline derivative, 4,5-methylenedioxy-1-pyridinedihydroisoquinoline (MPDQ), which contains a DNA intercalation moiety and a pyridine ring and can act as a chelating ligand (Scheme 1); the antitumor activity of Zn(II)/Ni(II) complexes has also been reported for the MPDQ ligand.36 As a part of our research for new metal-based anticancer agents, herein, we report the synthesis, crystal structure and in vitro antitumor activity of three copper(II) complexes with MPDQ as a chelating ligand; the mechanism of their cytotoxicity as well as their interactions with the potential target DNA were also investigated.
 |
| | Scheme 1 Isoquinoline ligand MPDQ. | |
Experimental
Materials and reagents
All chemicals, unless otherwise stated, were purchased from Sigma and Alfa Aesar. All materials were used as received without further purification unless stated otherwise. Tris–HCl–NaCl (TBS) buffer solution (5 mM Tris, 50 mM NaCl, pH adjusted to 7.35 by titration with hydrochloric acid using a Sartorius PB-10 pH meter, tris = tri(hydroxymethyl)aminomethane) was prepared using double-distilled water. The TBE buffer (1×) and DNA loading buffer (6×) were commercially available. Calf thymus DNA (ct-DNA) was purchased from Sino-American Biotech Co., Ltd. (Beijing, China). Ct-DNA gave a UV absorbance ratio at 260–280 nm of ∼1.85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, indicating that the DNA was effectively free of protein. The DNA concentration per nucleotide was determined spectrophotometrically by employing a molar absorption coefficient (6600 M−1 cm−1) at 260 nm. The stock solution of pUC19 plasmid DNA (250 μg mL−1) was purchased from Takara Biotech Co., Ltd.
:1, indicating that the DNA was effectively free of protein. The DNA concentration per nucleotide was determined spectrophotometrically by employing a molar absorption coefficient (6600 M−1 cm−1) at 260 nm. The stock solution of pUC19 plasmid DNA (250 μg mL−1) was purchased from Takara Biotech Co., Ltd.
Instrumentation
1H NMR and 13C NMR spectra were obtained on a Bruker AV-600 NMR spectrometer with DMSO-d6 as the solvent. Elemental analyses (C, H, and N) were carried out on a Perkin Elmer Series II CHNS/O 2400 elemental analyzer. ESI-MS spectra for the characterization of the complexes were performed on a Bruker HCT Electro spray Ionization Mass Spectrometer.
Synthesis of the copper(II) complexes
4,5-Methylenedioxy-1-pyridinedihydroisoquinoline (MPDQ) was synthesized according to previous reported methods.37
General procedures for the synthesis of complexes 1–3 are as follows. To a mixture of the corresponding metal salt, 4,5-methylenedioxy-1-pyridinedihydroisoquinoline (MPDQ), methanol (1 mL), and CHCl3 (0.5 mL) was placed in a thick Pyrex tube (∼20 cm long). The mixture was frozen by liquid N2 and evacuated under vacuum, then sealed with a torch, warmed to room temperature, and heated at 110 °C for several days to obtain the corresponding block crystals of complexes 1–3, suitable for X-ray diffraction analysis.
Complex 1. CuCl2·H2O (0.015 g, 0.1 mmol), MPDQ (0.050 g, 0.2 mmol). Yield 65%. Anal. found (%): C, 46.59; H, 3.13; N, 7.24. Calcd for: C, 45.98; H, 3.37; N, 7.16. ESI-MS: m/z 349.9 [Cu(MPDQ)Cl]+.
Complex 2. CuSO4·5H2O (0.025 g, 0.1 mmol), MPDQ (0.050 g, 0.2 mmol). Yield 70%. Anal. found (%): C, 41.91; H, 3.28; N, 6.52. Calcd for: C, 42.08; H, 3.21; N, 6.48. ESI-MS: m/z 412.4 [Cu(MPDQ)SO4]+.
Complex 3. Cu(ClO4)2·6H2O/(COOK)2·H2O (0.037 g/0.018 g, 0.1 mmol/0.1 mmol), MPDQ (0.050 g, 0.2 mmol). Yield 63%. Anal. found (%): C, 41.84; H, 2.63; N, 6.10. Calcd for: C, 41.92; H, 2.58; N, 6.17. ESI-MS: m/z 466.0 [Cu(MPDQ)ClO4 + H2O + CH3OH]+.
X-ray crystallography analysis
All reflection data were collected on an Agilent Super Nova diffractometer using graphite-monochromatic Mo-Kα radiation (λ = 0.71073 Å) at room temperature. The crystal structures were solved by the direct method using the program SHELXS-97 (ref. 38) and refined by the full-matrix least-squares method on F2 for all non-hydrogen atoms using SHELXL-97 (ref. 39) with anisotropic thermal parameters. All hydrogen atoms were located in calculated positions and refined isotropically, except for the hydrogen atoms of water molecules, which were fixed in a difference Fourier map and refined isotropically. The crystallographic data and refinement details of the structures are summarized in Table 1.
Table 1 Crystal data and structure refinement details for complexes 1–3
| Formula |
C30H24Cl4Cu2N4O6 |
C15H14CuN2O7S |
C32H24Cl2Cu2N4O16 |
| R1 = ∑ ‖Fo| − |Fc||/∑|Fo|. wR2 = [∑w(Fo2 − Fc2)2/∑w(Fo2). |
| Fw |
781.22 |
429.90 |
918.54 |
| T/K |
293(2) |
293(2) |
293(2) |
| Crystal system |
Triclinic |
Orthorhombic |
Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
Pbca |
P21/c |
| a, Å |
7.6582 (3) |
7.3674(1) |
7.8720(4) |
| b, Å |
8.8071 (4), |
18.0273(3) |
17.1458(5) |
| c, Å |
11.4566 (8) |
23.5080(4) |
12.8986(5) |
| α, ° |
97.269 (5) |
90.00 |
90.00 |
| b, Å |
101.942 (5) |
90.00 |
102.085(5) |
| γ, ° |
108.179 (4) |
90.00 |
90.00 |
| V, Å3 |
702.91 (6) |
3122.20(9) |
1702.35(12) |
| Z |
1 |
8 |
4 |
| Dc, g cm−3 |
1.846 |
1.769 |
1.792 |
| μ, mm−1 |
1.94 |
1.58 |
1.492 |
| GOF on F2 |
1.01 |
1.06 |
1.068 |
| Reflns (collected/unique) |
3194/199 |
11090/3236 |
17371/3478 |
| Rint |
0.023 |
0.021 |
0.0483 |
| R1a (I > 2σ(I)) |
0.0349 |
0.0266 |
0.0403 |
| wR2b (all data) |
0.0849 |
0.0718 |
0.0849 |
Cell lines, culture conditions and cytotoxicity assay
Human tumor cells BEL-7404, SK-OV-3, A549, A375, MGC-803 and NCI-H460 were obtained from the Shanghai Cell Bank in the Chinese Academy of Sciences. The tumor cells were grown in a RPMI-1640 medium supplemented with 10% (v/v) fetal bovine serum, 2 mM glutamine, 100 U mL−1 penicillin, and 100 U mL−1 streptomycin at 37 °C in a highly humidified atmosphere of 95% air/5% CO2. The cytotoxicity of the title complexes against BEL-7404, SK-OV-3, A549, A375, MGC-803 and NCI-H460 cells were examined by the micro culture tetrazolium (MTT) assay. The experiments were carried out using a reported procedure.40 The growth inhibitory rate of the treated cells was calculated using the data from three replicate tests as (ODcontrol − ODtest)/ODcontrol × 100%. The complexes were incubated with various cells for 48 h at five different concentrations of the complexes dissolved in fresh media; the range of concentrations used was dependent on the complexes. The final IC50 values were calculated by the Bliss method (n = 5). All tests were independently repeated at least three times.
Uptake of copper(II) complex in BEL-7404 and in nuclear DNA
The cells were seeded in a Petri dish. After 24 h of pre-incubation time in a drug-free medium at 37 °C in a humidified atmosphere of 5% CO2/95% air, complex 1 was added to give final concentrations of 10 and 20 μM. After a further 24 h of drug exposure, cell pellets were digested with concentrated HNO3, the resulting solutions were diluted with double-distilled water to a final concentration of 5% HNO3 (5 mL), and the copper contents were determined by plasma-mass spectrometry (ICP-MS). The data are the mean of three experiments and reported as mean ± SD.36
The cells were seeded in 6-well tissue culture plates for 24 h and then treated with complex 1 at 10 and 20 μM. After 24 h incubation, cells were washed three times with ice-cold PBS (1–2 mL). The genomic DNA was extracted according to the QIAamp DNA Mini Kit (50) protocol. Each DNA sample was digested with concentrated HNO3, the resulting solutions were diluted with double-distilled water to a final concentration of 5% HNO3 (5 mL), and the copper contents were determined by ICP-MS. The data are the mean of three experiments and reported as mean ± SD.36
Apoptosis assay by flow cytometry
The ability of copper complex 1 to induce apoptosis was evaluated in BEL-7404 cell lines using annexin-V conjugated with FITC and propium iodide (PI) counter staining by flow cytometry. BEL-7404 cells in exponential growth were incubated in 6-well plates and cultured for 12 h before the test compounds were added to the indicated final concentrations. After 48 h incubation, cells were harvested, washed twice in phosphate-buffered saline (PBS), and resuspended in 100 μL binding buffer (including 140 mmol L−1 NaCl, 2.5 mmol L−1 CaCl2 and 10 mmol L−1 Hepes/NaOH, pH 7.4) at a concentration of 1 × 106 cells mL−1. Then, the cells were incubated with 5 μL (5 μg mL−1) of annexin V–FITC (in buffer including 10 mmol L−1 NaCl, 1% bovine serum albumin, 0.02% NaN3 and 50 mmol L−1 Tris, pH 7.4) and 10 μL PI (20 μg mL−1) for 15 min at room temperature in the dark. Cells were kept shielded from light before being analyzed by flow cytometry using a Becton-Dickinson FACSCalibur.
Apoptosis assay by confocal microscopy
BEL-7404 cells were grown on chamber slides to 70% confluence. Complex 1 (0.5 μM) was added to the culture medium (final DMSO concentration, 5% v/v) and incubated for 12 h at 37 °C. The cells were then washed with PBS, stained with a medium containing DAPI/DiD solution (100 mg mL−1 DAPI, 100 mg mL−1 DiD) for 30 min and photographed with a Zeiss LSM7.
Cell cycle analysis
BEL-7404 cell lines were maintained in Dulbecco's modified Eagle's medium with 10% fetal calf serum in 5% CO2 at 37 °C. Cells were harvested by trypsinization and rinsed with PBS. After centrifugation, the pellet (105 to 106 cells) was suspended in 1 mL of PBS and kept on ice for 5 min. The cell suspension was then fixed by the dropwise addition of 9 mL pre-cooled (4 °C) 100% ethanol with violent shaking. Fixed samples were kept at 4 °C until use. For staining, cells were centrifuged, resuspended in PBS, digested with 150 mL of RNase A (250 mg mL−1) and treated with 150 mL of propidium iodide (PI) (0.15 mM), then incubated for 30 min at 4 °C. PI-positive cells were counted with a FACScan Fluorescence-activated cell sorter (FACS). The population of cells in each cell cycle phase was determined using Cell Modi FIT software (Becton Dickinson).
Determination of caspase-3 activity
The activation of caspase-3 was examined using a caspase-3 (DEVD-FMK) conjugated to FITC as the fluorescent in situ marker in living cells (catalog#QIA91, Merk). FITC–DEVD-FMK is cell-permeable, nontoxic, and irreversibly binds to activated caspase-3 in apoptotic cells. After exposure of BEL-7404 cell lines to complex 1 for 4 h, the cultures were washed twice with HBSS containing 0.1% BSA and then incubated with FITC–DEVD-FMK (1 μL mL−1 in EBSS) for 1 h in a 37 °C incubator with 5% CO2. The FITC label in apoptotic cells was examined immediately under a fluorescence microscope with excitation and emission at 485 and 535 nm, respectively.
Mitochondrial membrane potential detection
Cells incubated with the IC50 concentration of complex 1 for 12 h in poly-HEMA coated 6-wells were collected and resuspended in a fresh medium. After the addition of 0.5 mL JC-1 working solution, the cells were incubated in a 5% CO2 incubator for 20 min at 37 °C. The staining solution was removed by centrifugation and cells were washed with JC-1 staining buffer twice. Cells having low membrane potential were photographed under a fluorescent microscope (120×).
Western blotting
BEL-7404 cells (5 × 105) were cultured on a 60 mm dish and incubated overnight before experiments were carried out, which involved treatment with complex 1 at a concentration of 5 μM for 12, 24 and 36 h. After incubation, cells were harvested and lysed using a lysis buffer (150 mM NaCl, 100 mM Tris–HCl, pH 7.4, 10% glycerol, 1% Triton X-100, 10 mM NaF, 5 mM sodium pyrophosphate, 5 mM sodium orthovanadate, 0.1% SDS) with a protease inhibitor. Total protein extracts (50 μg) were loaded onto a suitable concentration of SDS-polyacrylamide gel and were then transferred to polyvinylidene fluoride (PVDF) membranes. The membrane was blocked with 5% BSA in TBST buffer and incubated with the corresponding primary antibodies at 4 °C overnight; P-53 (ab17990, 1:500), Bcl-2 (ab32124) (1:2000), cleaved caspase-3 (ab32042) (1:500) and cleaved caspase-9 (ab32539) (1:500) were obtained from Abcam. After washing, the membrane was incubated with a secondary antibody conjugated with horseradish peroxidase (1:2500) for 120 min. The immuno reactive signals were detected using an enhanced chemoluminance kit (Pierce ECL Western Blotting Substrate) following the procedures given in the user manual.
DNA binding studies
The 2 × 10−3 M ct-DNA stock solution was stored at 4 °C for not more than 5 days before use. The synthetic copper(II) complexes were all prepared as 2 × 10−3 M DMSO stock solutions for DNA binding studies. The final working solutions of the complexes for DNA binding studies were diluted by TBS and the DMSO content was less than 10%. A solution containing 2 × 10−4 M DNA and 2 × 10−5 M EB ([DNA]/[EB] = 10:1) was prepared for EB–ct-DNA competitive binding studies. The melting temperatures of DNA and the DNA-complexes were determined by monitoring the absorbance at 260 nm of the samples at different temperatures. The absorbance intensities were then plotted as a function of the temperature ranging from 25 to 95 °C. The values of the melting temperatures of DNA and the DNA-complexes were obtained from the transition midpoint of the melting curves based on fss versus temperature (T), wherein fss = (A − A0)/(Af − A0), where A0 is the initial absorbance intensity, A is the absorbance intensity corresponding to its temperature, and Af is the final absorbance intensity.41 Fluorescence emission spectra were obtained under slit width as 10 nm/10 nm for Ex/Em, respectively. The quenching constant for comparing the efficiency of fluorescence quenching, i.e., Kq of each compound was obtained by the linear fit of a plot of I0/I versus [Q], according to the classic Stern–Volmer equation: I0/I = 1 + Kq × [Q],42 where I0 and I are the peak emission intensities of the EB–ct-DNA system in the absence and presence of each compound as a quencher, and [Q] is the concentration of the quencher. CD absorption spectra of DNA were obtained in TBS at a 100 nm min−1 scan rate in the wavelength range from 200 to 400 nm with 1 × 10−4 M DNA in the absence and presence of each compound at 2 × 10−5 M. The CD signal of TBS was taken as the background and subtracted from the spectra. All the spectroscopy experiments were performed at 25 °C. In DNA viscosity measurements, ct-DNA was dissolved in BPE buffer (6 mM Na2HPO4, 2 mM NaH2PO4, 1 mM Na2EDTA, pH 1/4 7.2) to prepare an 8 × 10−4 M working solution. The compounds were added until the desired [compound]/[DNA] ratios were reached, ranging from 0.02 to 0.20 in every 0.02 interval. The circulating bath temperature was maintained at 35.0 ± 0.1 °C. The viscosities η (unit: cP) were directly obtained by running spindle in the working samples at 30 rpm. The data were presented as η/η0 versus [compound]/[DNA] ratio, in which η0 and η refer to the viscosity of the DNA working solutions in the absence and presence of the corresponding compound, respectively. The influence of the solvent DMSO on the viscosity was eliminated by subtracting its viscosity 2.03 cP (measured at 35.0 °C) from the sample viscosities.
Statistics
The data processing included the Student's t-test with P ≤ 0.05 taken as significance level, using SPSS 13.0.
Results and discussion
Synthesis
The isoquinoline ligand MPDQ was prepared according to the method reported in our prior study, which was synthesized by Bischler-Napieralski cyclization of the acylation product (ALP) of phomopiperonylamine and picolinic acid (Scheme 1).37
Jan Reedijk's works indicated that metal complexes with different anions and identical ligands had different bioactivities;43 herein, the present three Cu(II) complexes [Cu2(MPDQ)2Cl4] (1), [Cu(MPDQ)(H2O)(SO4)] (2), and [Cu2(MPDQ)2(C2O4)(ClO4)2] (3) were obtained by treating MPDQ with CuCl2·2H2O, CuSO4·5H2O and Cu(ClO4)2·6H2O/(COOK)2·H2O (1:1) in CH3OH/CHCl3 (2:1) under solvothermal conditions.
The products were characterized by elemental analysis, ESI-MS spectroscopy and single crystal X-ray diffraction analysis.
Structure description and stability in solution
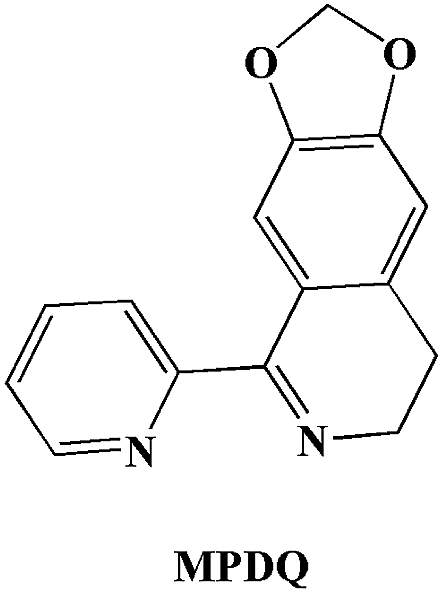
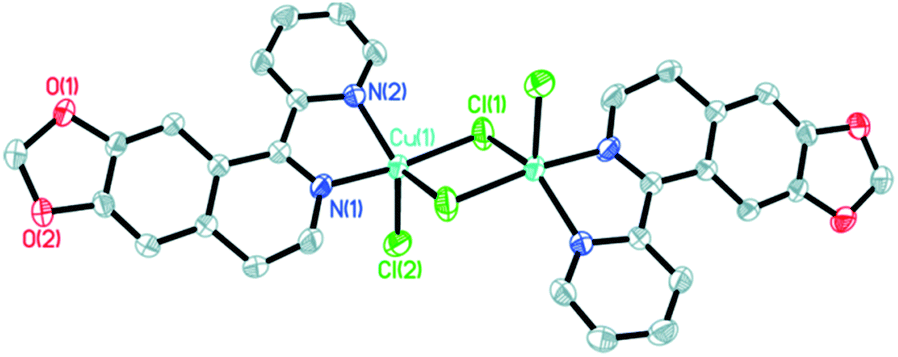
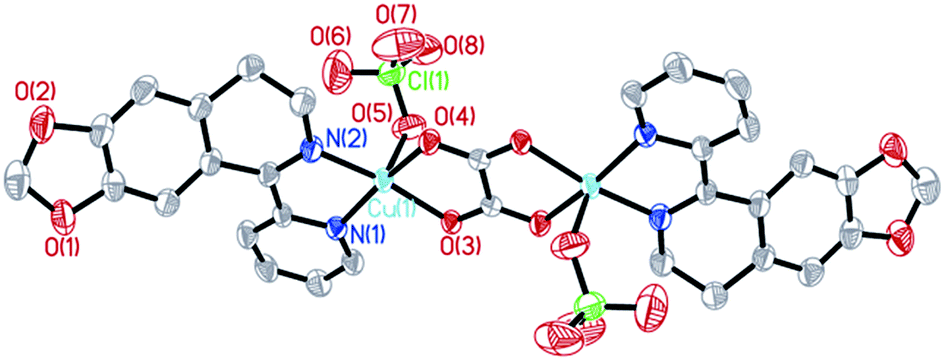
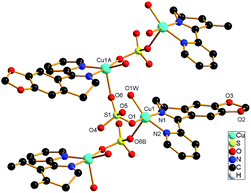
The molecular structures of the three complexes [Cu2(MPDQ)2Cl4] (1), [Cu(MPDQ)(H2O)(SO4)] (2), and [Cu2(MPDQ)2(C2O4)(ClO4)2] (3) are depicted in Fig. 1–3. Complexes 1 and 3 are binuclear structures, whereas complex 2 is a one-dimensional chain structure. As shown in Fig. 1, the two Cu(II) centers in complex 1 are bridged by two chloride anions. Each Cu(II) centre adopts a distorted square pyramidal geometry by coordinating with one Cl, two μ2-Cl, and two N atoms from MPDQ. As shown in Fig. 2, the Cu(II) centre in complex 2 adopts a distorted square planar geometry by coordinating with one aqua, one O atom from the μ2-SO42− anion, and two nitrogen atoms from MPDQ. By means of μ2-SO42− linking two adjacent copper(II) centers, a one-dimensional chain structure is formed. Complex 3 is a dinuclear structure, in which the [Cu(MPDQ)(ClO4)]+ cation is bridged by one tetra-dentate oxalate anion; the Cu(II) centre adopts a distorted square pyramidal geometry by coordinating with one O from ClO4−, two oxygen from the oxalate anion, and two nitrogen from MPDQ. Similarly, two chlorine atoms in complex 1 and two perchlorates in complex 3 lie in the opposite sides of the plane formed by bridged μ2-Cl or oxalate and two copper(II) centers. For the reported [Zn2(L)2(μ2-Cl)2Cl2] complex,37 the ESI-MS results show that the chlorine bridge of complex 1 is broken in a polar solution system (DMSO, methanol, H2O) and exists in a mononuclear form, [CuCl(MPDQ)]+ (ESI-MS: m/z [CuCl(MPDQ)]+ = 349.9). As with complex 1, complexes 2 and 3 have also been dissociated into a mononuclear form in a polar solution system (DMSO, methanol, H2O), ESI-MS: m/z 412.4 [Cu(MPDQ)Cl]+ (for complex 2), and m/z 466.0 [Cu(MPDQ)ClO4+H2O + CH3OH]+ (for complex 3). The stabilities of the Cu complexes were investigated under physiological conditions (Tris–KCl–HCl buffer solution with a pH value of 7.35, containing 1% DMSO) by means of HPLC. As shown in Fig. S1 (ESI†), the time-dependent (at 12, 24, and 48 h) HPLC spectra of each complex indicated that the present Cu(II) complexes were stable in TBS (Tris–KCl–HCl) buffer solution for 48 h at room temperature.
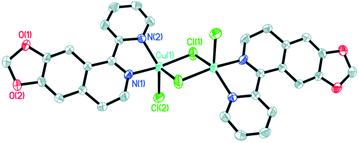 |
| | Fig. 1 ORTEP drawing of complex 1. The hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°) for complex 1: Cu(1)–N(1) 1.9713(22), Cu(1)–N(2) 2.0433(20), Cu(1)–Cl(1) 2.2718(7), Cu(1)–Cl(2) 2.3480(8), N(1)–Cu(1)–N(2) 78.87(8), Cl(1)–Cu(1)–N(1) 70.69(8), Cl(2)–Cu(2)–N(1), 92.06(8), N(2)–Cu(1)–Cl(1) 96.17(6), N(2)–Cu(1)–Cl(2) 122.61(6), Cl(1)–Cu(1)–Cl(2) 97.25(3). | |
 |
| | Fig. 2 ORTEP drawing of complex 2. The hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) angles (°), for complex 2: Cu(1)–O(1w) 1.9576(15), Cu(1)–O(1) 1.9620(14), Cu(1A)–O(6) 2.2221(15), Cu(1)–N(1) 1.9648 (17), Cu(1)–N(2) 1.9944(17); O(1)–Cu(1)–O(1w) 93.81(6), N(1)–Cu(1)–O(1w) 91.83(7), N(2)–Cu(1)–O(1w) 159.85(7), N(1)–Cu(1)–O(1) 172.25 (7), N(2)–Cu(1)–O(1) 92.32(7), N(2)–Cu(1)–N(1) 80.62(7). (Symmetry code A: 1/2 + x, 1/2 − y, −z; B: −1/2 + x, 1/2 − y, −z). | |
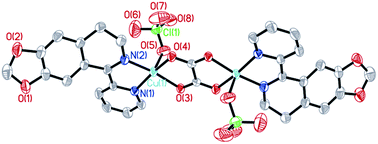 |
| | Fig. 3 ORTEP drawing of complex 3. The hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) angles (°): Cu(1)–N(1) 1.9693(23), Cu(1)–N(2) 1.9483(25), Cu(1)–O(3) 1.9744(19), Cu(1)–O(4) 1.9917(19), Cu(1)–O(5) 2.2616(27), N(1)–Cu(1)–N(2) 82.27(10), O(3)–Cu(1)–N(2) 169.66(10), O(4)–Cu(2)–N(2) 95.80(9), O(5)–Cu(1)–N(2) 103.60(11), N(1)–Cu(1)–O(3) 95.04(9), N(1)–Cu(1)–O(4) 166.73(11), N(1)–Cu(1)–O(5) 96.51(11), O(3)–Cu(1)–O(4) 84.52(8), O(3)–Cu(1)–O(5) 86.60(9), O(4)–Cu(1)–O(5) 96.70(10). | |
In vitro cytotoxic activity
The in vitro cytotoxicity of MPDQ and complexes 1–3 against BEL-7404, SK-OV-3, A549, A375, MGC-803, NCI-H460 and HL-7702 cell lines were investigated (using cisplatin as the positive control). As shown in Table 2, towards the tested cells, IC50 exhibited significantly enhanced activities compared to that of free MPDQ and the corresponding copper(II) salts, suggesting a synergistic effect between MPDQ and copper(II) ions. It should be noted that these copper(II) complexes displayed lower cytotoxicity towards normal human liver HL-7702 cells than towards the tested cancer cells, thereby, complexes 1–3 exhibited selectivity to cancer cells to a certain extent. Among these, complex 1 exhibited the highest cytotoxicity against BEL-7404, A549 and A375 with IC50 values of 1.41–3.34 μM. Complex 3 also exhibited higher cytotoxicity against BEL-7404, A549 and MGC-803 with IC50 values of 3.39–6.32 μM. Based on their structures and the molecular species in solution (ESI-MS data), it could be envisioned that the approximately planar benzo[1,3]dioxole moiety of MPDQ that can intercalate between adjacent base pairs of DNA, together with the copper ion, played a key role in their cytotoxicity.31 Since complex 1 displayed the highest cytotoxicity in most cases, this was used as a representative compound in the following studies on the mechanism of cell cytotoxicity against the BEL-7404 cancer cell.
Table 2 IC50 (μM) values for complexes 1–3 against six human tumor cell lines
| Complexes |
BEL-7404 |
SK-OV-3 |
A549 |
A375 |
MGC-803 |
NCI-H460 |
HL-7702 |
| Cisplatin was used as a positive control. ND is no data. |
| MPDQ |
38.41 ± 0.27 |
67.58 ± 4.87 |
31.55 ± 3.46 |
37.67 ± 2.39 |
55.41 ± 1.58 |
>100 |
>100 |
| 1 |
3.34 ± 0.51 |
18.27 ± 0.14 |
1.41 ± 0.25 |
2.38 ± 0.04 |
14.57 ± 2.11 |
34.54 ± 3.07 |
20.78 ± 0.09 |
| 2 |
11.25 ± 0.21 |
24.76 ± 0.32 |
14.24 ± 0.18 |
8.29 ± 0.22 |
7.48 ± 0.34 |
24.81 ± 1.25 |
68.52 ± 0.06 |
| 3 |
4.12 ± 0.07 |
17.33 ± 0.41 |
6.32 ± 0.08 |
11.57 ± 0.36 |
3.39 ± 0.06 |
17.23 ± 0.78 |
28.66 ± 0.16 |
| CuCl2·2H2O |
74.38 ± 5.68 |
NDb |
81.31 ± 2.17 |
60.26 ± 3.04 |
72.15 ± 3.17 |
41.07 ± 1.54 |
>100 |
| CuSO4·5H2O |
>100 |
NDb |
NDb |
>100 |
>100 |
82.44 ± 3.37 |
70.10 ± 0.06 |
| Cu(ClO4)2·6H2O |
63.26 ± 4.14 |
NDb |
87.54 ± 6.22 |
69.13 ± 1.12 |
52.24 ± 2.29 |
53.19 ± 4.10 |
>100 |
| Cisplatina |
64.22 ± 1.21 |
84.21 ± 0.24 |
17.21 ± 0.58 |
78.54 ± 2.25 |
23.58 ± 0.43 |
48.52 ± 0.32 |
74.25 ± 0.11 |
The uptake of copper(II) complex in BEL-7404 cells and nuclear DNA
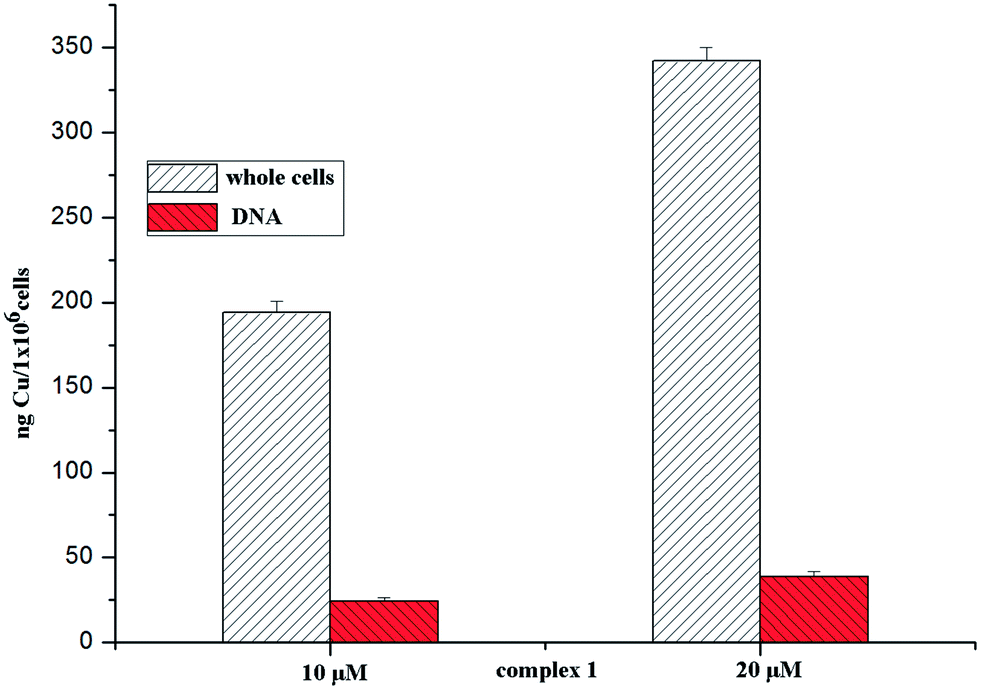
The cellular uptake properties of metal-based anticancer drugs are important factors that can influence their antiproliferative efficacies. To investigate whether copper complexes enter cells, we determined the copper uptake of complex 1 (which has lower IC50 values) in BEL-7404 cells by ICP-MS. Cells were treated with complex 1 at 10 and 20 μM for 24 h and the accumulations of copper in the 106 cells were 1940 ng and 3427 ng, respectively (see Fig. 4.). The results indicated that complex 1 could penetrate the cell membrane well.
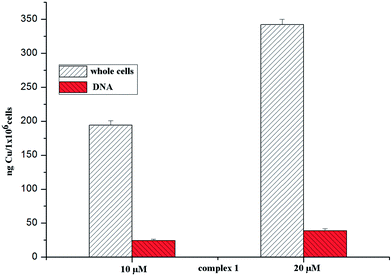 |
| | Fig. 4 The uptake and DNA accumulation of elemental copper determined in BEL-7404 cells after 48 h of exposure to 10/20 μM of complex 1. | |
To verify the supposition that DNA may be an important target for the complexes, the copper contents of complex 1 in the DNA sample that was isolated have been examined by ICP-MS. After 24 h of exposure to complex 1 at 10 and 20 μM, the uptake of elemental copper by the DNA samples was 13.7 ng and 26.4 ng, respectively; the results are shown in Fig. 4. This suggested that complex 1 could be transported across the cellular phospholipid membrane, finally combining and then interacting with the DNA.
Apoptosis study by flow cytometry and confocal microscopy
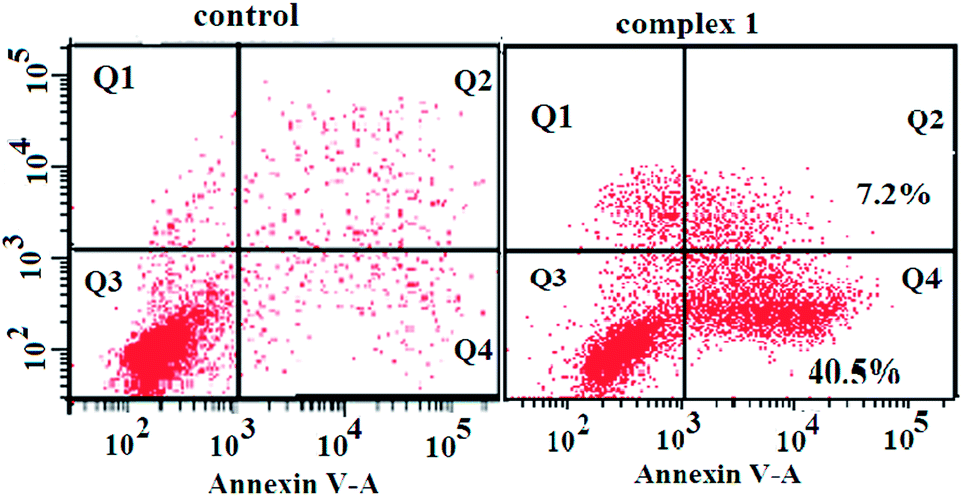
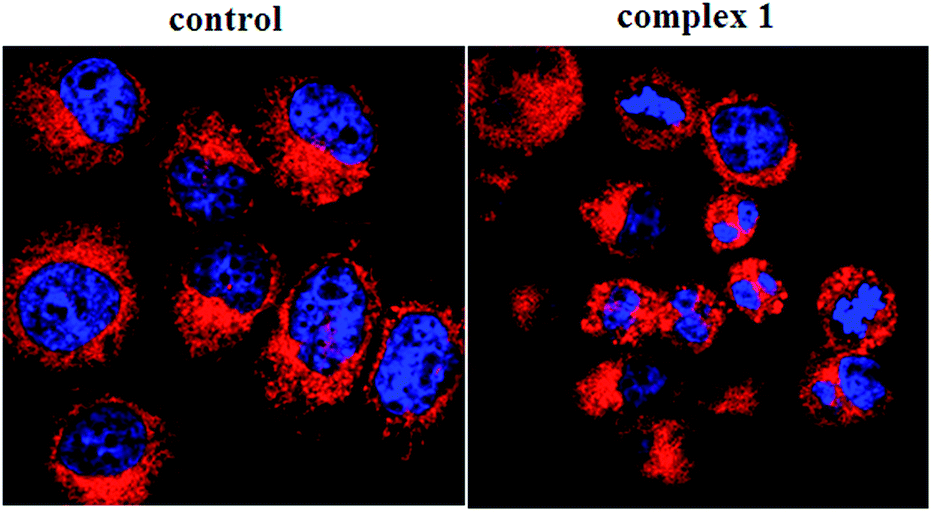
To determine whether the observed cell death induced by the complexes was due to apoptosis or necrosis, the interaction of BEL-7404 cells with complex 1 was firstly investigated using an annexin V–FITC/propidium iodide assay. As phosphatidylserine (PS) exposure usually precedes the loss of plasma membrane integrity in apoptosis, the presence of annexin V+/PI− cells is considered an indicator of apoptosis. As shown in Fig. 5, in the presence of complex 1, the population of annexin V+/PI− cells (Q4) was 40.5%, which suggested that apoptotic death was induced in BEL-7404 cells. On the basis of cell morphology and cell membrane integrity, normal, necrotic, and apoptotic cells can be distinguished by laser confocal microscopy. Apoptosis induction by complex 1 was further confirmed by a DAPI/DiD staining assay. As shown in Fig. 6, from which the characteristic morphological changes under the treatment of complex 1 can be observed, the nucleus of a living cell was stained as green fluorescence and red fluorescence represented the cell membrane. In the presence of complex 1, damage to the cell membrane and the apoptotic bodies of the nucleus were observed. These results indicated that the cell death induced by complex 1 was mainly caused by the apoptotic pathway.
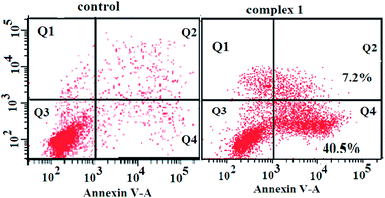 |
| | Fig. 5 Annexin-V/propidium iodide assay of BEL-7404 cells treated by complex 1 (at IC50 concentration) measured by flow cytometry. | |
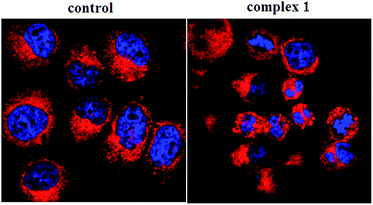 |
| | Fig. 6 DAPI/DiD assay of BEL-7404 cells treated by complex 1 (at IC50 concentration) measured with a confocal microscope. | |
Cell cycle arrest
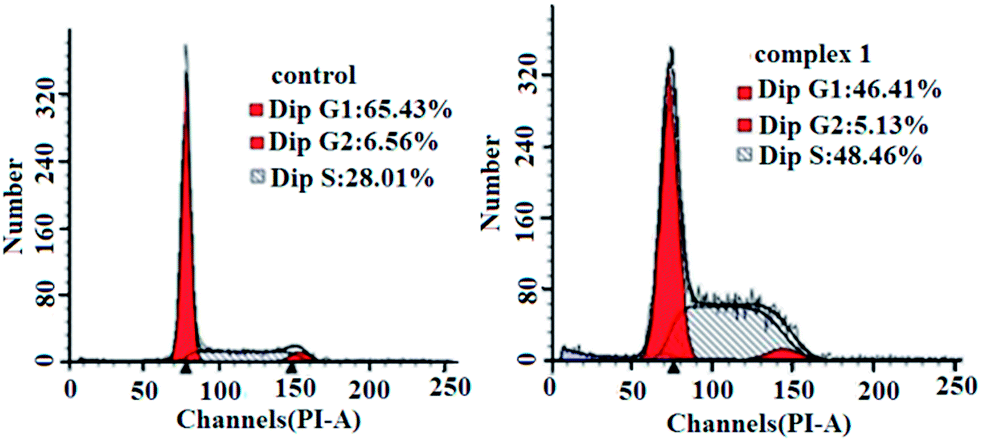
To determine whether the suppression of cancer cell growth by the complexes was caused by a cell cycle arrest, BEL-7404 cells were treated with complex 1 at IC50 concentrations and the cell cycle phase was assayed by assessing the DNA content of cells stained with propidium iodide as measured by flow cytometry. As shown in Fig. 7, complex 1 caused increased accumulation of BEL-7404 cells in the S phase and a decrease in the accumulation in the G0/G1 phase of the cell cycle. The cell population of the S phase increased from 28.01% to 48.46% in the BEL-7404 cells, compared with the control; complex 1 induced a drastic cell population decrease in the G1 phase (from 65.43% to 46.41%) along with a concomitant accumulation in the S phase in BEL-7404 cells. These results indicated a remarkable S phase arrest. Actually, most DNA replication occurs within this stage; therefore, it could be concluded that DNA may be one of the intracellular targets for complex 1.
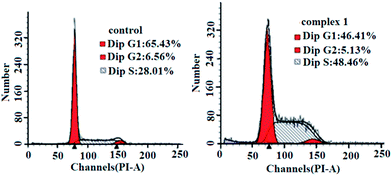 |
| | Fig. 7 Cell cycle analysis by flow cytometry for BEL-7404 cells treated with complex 1. | |
Detection of mitochondrial membrane potential
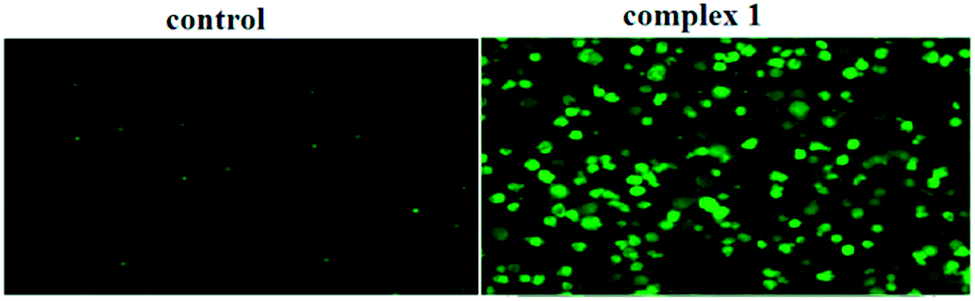
Mitochondria act as a point of integration for apoptotic signals, originating from both the extrinsic and intrinsic apoptotic pathways. Mitochondrial dysfunction and the release of apoptogenic factors are critical events in triggering various apoptotic pathways. Loss of membrane potential is an important indicator of mitochondrial dysfunction. To delineate the importance of mitochondria in complex 1-induced BEL-7404 apoptosis, variations in the mitochondrial membrane potential were measured using the JC-1 probe, which was dispersed from the aggregated form (red fluorescence) to the monomeric form (green fluorescence) when the mitochondrial membrane potential was lost. As shown in Fig. 8, the BEL-7404 cells treated with complex 1 exhibited a significant decrease in mitochondrial membrane potential as indicated by a notable shift in the ratio of green/red fluorescence versus the control. The results suggested that mitochondria are involved in the process of apoptosis.
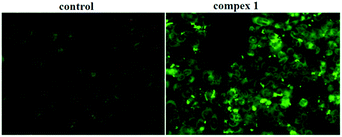 |
| | Fig. 8 Loss of ΔΨm induced by complex 1. Cells were treated with complex 1 for 0.5 h and examined by JC-1 under a fluorescence microscope. | |
Caspase-3 activation assay
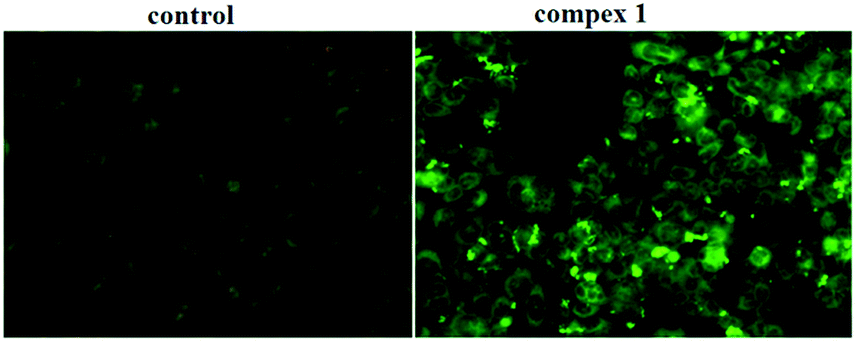
It is well known that the caspase family plays a crucial role in the modulation of programmed cell death (PCD), which is a genetically regulated, evolutionarily conserved process with numerous links to many human diseases, most notably cancer. Caspase-3, the executioner caspase, is able to directly degrade multiple substrates, including structural and regulatory proteins. Thus, therapeutic strategies designed to stimulate apoptosis by activating caspase-3 may combat cancer. Some small molecules have been shown to be selective activators of caspase-3; therefore, we investigated whether caspase-3 is activated when BEL-7404 cells are exposed to complex 1. As shown in Fig. 9, the BEL-7404 cells treated with complex 1 displayed a significant increase in the activity of caspase-3 as indicated by a notable shift in the ratio of green/dark fluorescence versus the control. These results suggested that complex 1 could be an efficient activator of caspase-3.
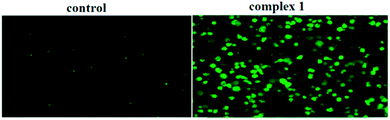 |
| | Fig. 9 The activation of caspase-3 induced by complex 1, examined by FITC–DEVD-FMK under a fluorescence microscope. | |
The measurement of intrinsic pathway
The intrinsic pathway (via mitochondria) plays a key role in regulating apoptosis in response to various stimuli, beginning with the upregulation of wild-type p53 followed by changes in Bcl-2 and Bax, which increase the permeabilization of the mitochondrial outer membrane, allowing alteration of the mitochondrial membrane potential, which is also mediated by the release of cytochrome-c (Cyt-c), Ca2+ from mitochondria. Cyt-c then binds in a complex with Apaf-1 and dATP, followed by activation of caspase-9 and caspase-3, then multiple substrates, including structural and regulatory proteins, are degraded and the apoptosis of cells occurs.44 The examination of mitochondrial membrane potential and caspase-3 activation indicated that the intrinsic pathway is involved in the apoptotic process of BEL-7404 cells induced by complex 1. To further confirm the intrinsic apoptotic pathway activated by complex 1, we analyzed the expression levels of a series of key proteins that are related to the intrinsic pathway by means of Western blotting (see Fig. 10). The studies showed that P53 proteins were up-regulated and Bcl-2 proteins were down-regulated; at the same time, both caspase-9 and caspase-3 proteins were activated over a prolonged time at 5 μM. The examination of four proteins further confirmed that the apoptosis of BEL-7404 cells induced by complex 1 was related to the intrinsic pathway.
 |
| | Fig. 10 Western blot analysis: the expression of a series of proteins involved in mitochondria-mediated apoptosis after treatment with complex 1 for the indicated concentration and times. | |
DNA binding study
Despite the presence of other biological targets in tumor cells, including RNA, enzymes and protein, it is generally accepted that DNA is the primary target for many metal-based anticancer drugs such as cisplatin.45 Our previous study indicated that the metal complexes of isoquinoline alkaloids might act as bifunctional DNA-binding agents, in which the isoquinoline ligand intercalates between the neighboring base pairs of DNA and the metal ions, such as platinum(II) and zinc(II), covalently bind to DNA.29–31 The ICP-MS and cell cycle arrest experiments indicated that the DNA may be one important target of complex 1; in order to investigate the DNA binding properties of our newly-synthesized isoquinoline copper complexes, a series of experiments exploring DNA binding were carried out, including DNA-melting analysis, fluorescence competitive binding, CD analysis, viscosity measurements and agarose gel electrophoresis assay.
DNA-melting analysis
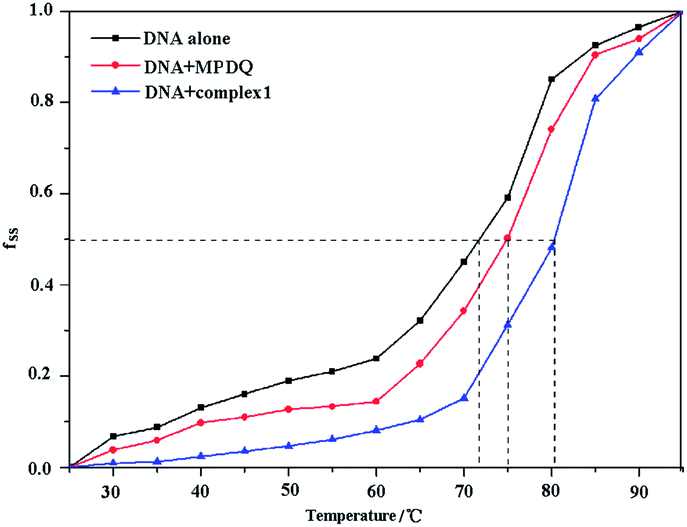
The melting temperature (Tm) of DNA, which is defined as the temperature at which half of the dsDNA is dissociated into single strands, is strictly related to the stability of the macromolecule. In addition, the melting temperature of DNA may be altered by interaction with chemicals. In general, the classic intercalation of small molecules into the double helix can increase the stability of the DNA helical structure and lead to an increase of 5–8 °C in the Tm of DNA, while non-intercalative binding causes no visible enhancement of the Tm.46 The melting curves of DNA and the DNA compounds systems are shown in Fig. 11 and S2.† As seen from the figures, the Tm values of DNA in the absence and presence of the compounds were 71.97 °C, 75.03 °C (MPDQ), 80.26 °C (complex 1), 77.10 °C (complex 2) and 79.35 °C (complex 3), indicating that the stability of DNA increased in the presence of the compounds. The Tm values of DNA in the presence of MPDQ only increased by about 3.06 °C; in contrast, larger increases in the Tm values of DNA can be achieved in solution with copper(II) complexes, which confirms that stronger intercalative binding of copper complexes to DNA occurred. Although the Tm values of DNA in the presence of copper complexes exhibited a greater increase than with MPDQ, the intercalative binding capacity of the copper(II) complexes was weaker than that of EtBr, because EB induced an increase of 13 °C in the Tm of DNA.47
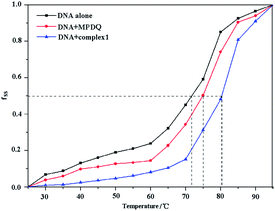 |
| | Fig. 11 The melting curves of DNA in the absence and presence of complex 1 and MPDQ. | |
Competitive binding study
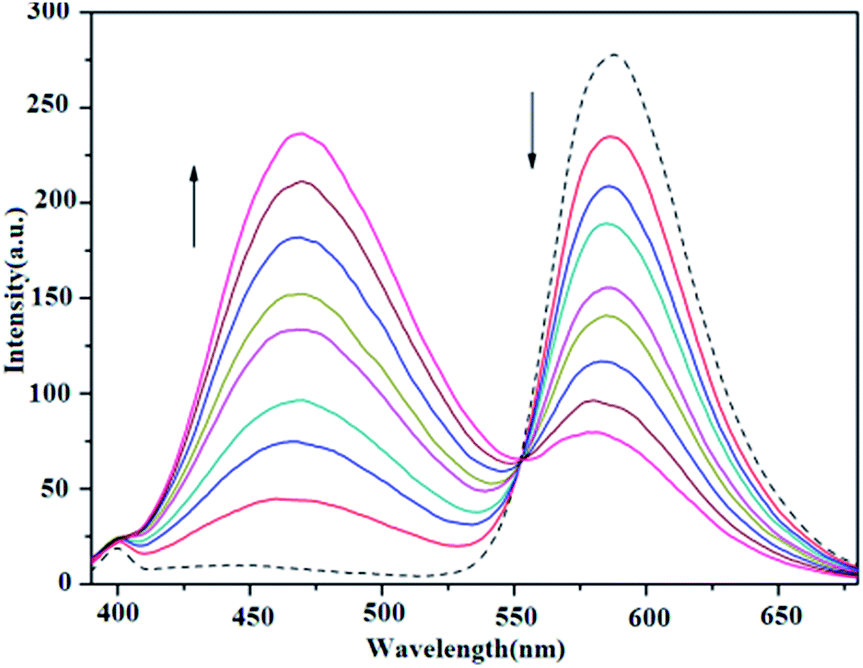
The binding of all the present copper complexes and MPDQ to ct-DNA were investigated by a competing assay with ethidium bromide (EB) as an intercalative agent.40 In the competitive binding experiments, the EB–ct-DNA system with [EB]/[DNA] = 1:10 ([EB] = 1 × 10−5 M, [DNA] = 1 × 10−4 M) showed a characteristic strong emission at ca. 588 nm when excited at 332 nm, indicating that the intercalated EB molecules were sufficiently protected by the neighboring base pairs in the ct-DNA from being quenched by polar solvent molecules. When increasing the [compound]/[EB] ratio in the copper complexes from 1:1 to 8:1, the characteristic emission of EB decreased, especially for copper(II) complexes 1 and 3 (as shown in Fig. 12 and S5†). Table 3 lists the corresponding data with respect to EB fluorescence quenching and the calculated competitive binding ability. It was found that all the complexes could quench the fluorescence emission of EB to a certain extent, which suggested that they may compete with EB to bind to ct-DNA through intercalation at similar binding sites.48 Furthermore, the quenching constant Kq was also calculated by restricted linear fitting of [I0/I] to [Q] from the Stern–Volmer equation and is listed in Table 3. The results indicated that copper(II) complexes 1 and 3 exhibited a stronger quenching ability but complex 2 and the ligand MPDQ were weaker.
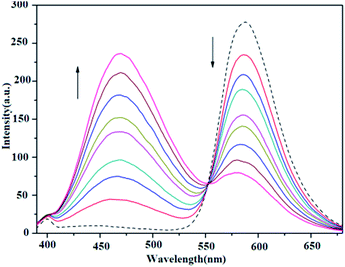 |
| | Fig. 12 Fluorescence emission spectra of EB bound with ct-DNA in the absence (dashed line ---) and presence (solid lines —) of complex 1 as competitive agent with increasing [complex 1]/[EB] ratios from 1:1 to 8:1. | |
Table 3 Calculated quenching ratios of EB by titration of complexes to EB–ctDNA system
| Complexes |
One-step FI quenching ratioa |
Total FI quenching ratio |
Kq |
| FI, fluorescence intensity; one-step refers to [compound]/[EB] = 1:1. |
| Complex 1 |
15.67% |
72.55% at [complex 1]/[EB] = 8:1 |
0.33 × 104 |
| Complex 2 |
1.53% |
18.37% at [complex 1]/[EB] = 8:1 |
0.11 × 103 |
| Complex 3 |
13.78% |
66.74% at [complex 1]/[EB] = 8:1 |
0.27 × 104 |
| MPDQ |
0.52% |
5.85% at [MPDQ]/[EB] = 8:1 |
0.51 × 102 |
Circular dichroism (CD) analysis
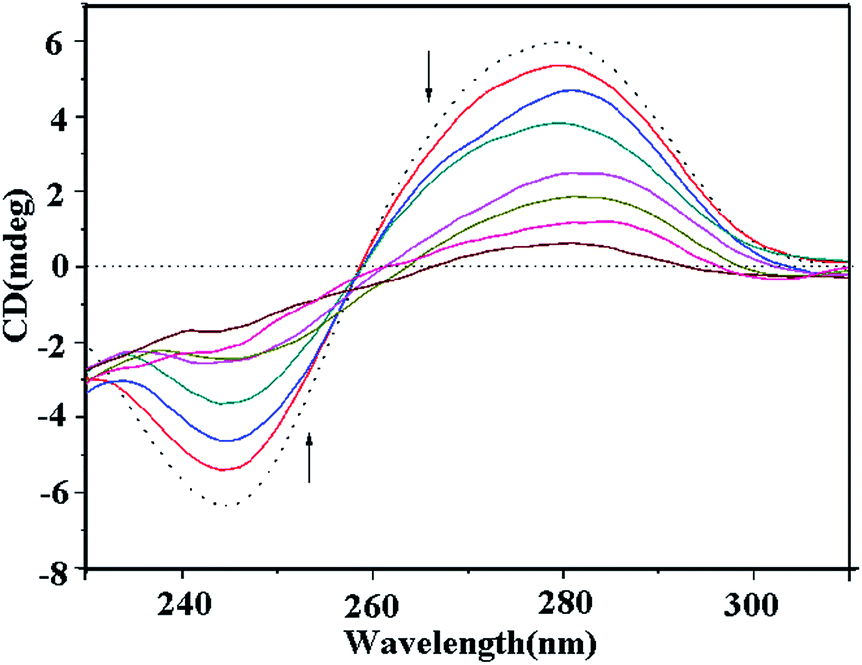
At [DNA]/[compound] ratios from 10:1 to 10:7, the presence of copper complex 1 induced an obvious decrease in the positive and negative absorption of ct-DNA, which represents the intercalative binding of complex 1 with DNA (Fig. 13).49,50 At the same concentration, complexes 2 and 3 also induced significant changes in both the positive and negative absorption (Fig. S7 and 8†); however, there were fewer variations in the CD characteristic DNA absorption peaks. These results indicated that all the complexes existed in an intercalative binding mode with DNA but the intercalation of the copper(II) complex 1 with DNA was still stronger than that of the ligand MPDQ and complexes 2 and 3.
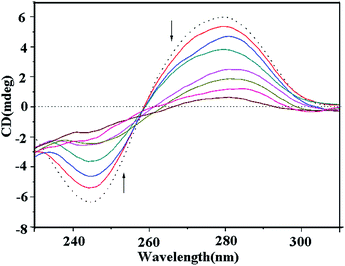 |
| | Fig. 13 Circular dichroism spectra of ct-DNA bound by complex 1 with [DNA]/[compound] ratios from 2:1 to 7:1 (DNA alone of 1 × 10−4 M, dashed line; DNA bound by compounds, colored solid lines). | |
Viscosity measurement studies
When small planar molecules intercalate into the neighboring base pairs of DNA, the DNA double helix loosens to accommodate intercalation, resulting in a lengthening of the DNA helix. Since the viscosity of the DNA solution is very sensitive to changes in the length of DNA, the viscosity of the DNA solution is usually increased in the intercalative binding mode as a result of an increase in the length of DNA.51
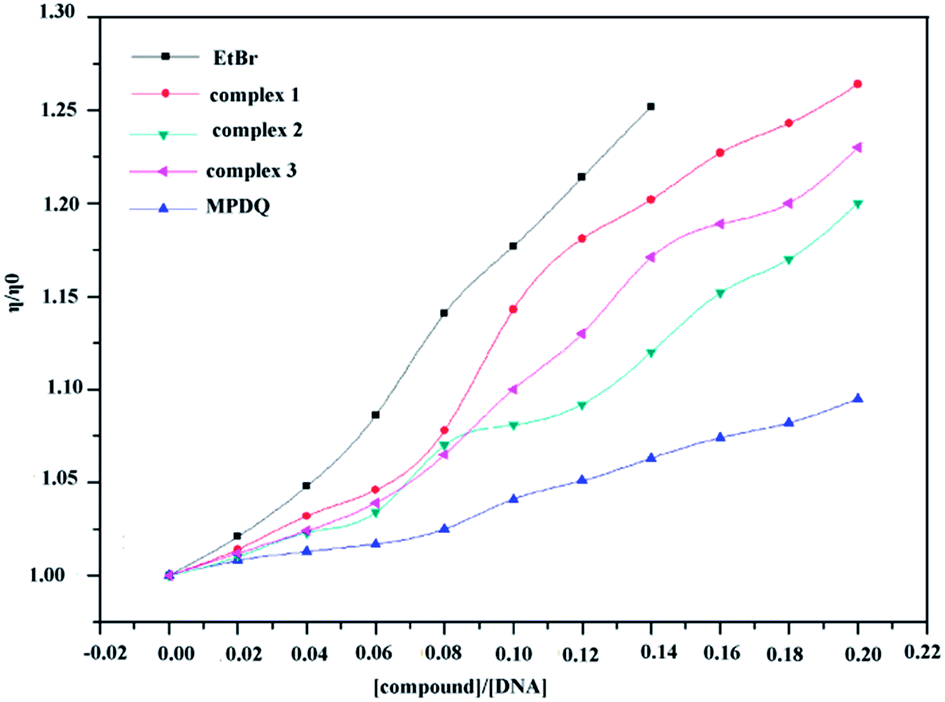
The influences of binding copper(II) complexes and MPDQ with ct-DNA on the viscosity of the ct-DNA solution are shown in Fig. 14, in which EtBr was employed as the intercalative binding indicator. The addition of MPDQ resulted in an increase in the viscosity of the DNA solution but to a very small extent; the DNA viscosity only increased by about 8.1% as the [MPDQ]/[DNA] ratio increased from 0.02 to 0.20. In contrast, as the [copper(II) complexes]/[DNA] ratio increased from 0.02 to 0.20, the viscosity of all the DNA solutions showed a more obvious increase than with MPDQ. Based on the above mentioned results, it can be concluded that an intercalative binding mode exists between the copper(II) complexes and ct-DNA; furthermore, among all the copper(II) complexes, complex 1 induced the greatest viscosity increase in ct-DNA solutions. Nevertheless, the intercalative binding capacity of copper(II) complex 1 was still somewhat weaker than that of EtBr. EtBr induced an increase in the DNA viscosity of 24.81% at a [EtBr]/[DNA] ratio of only 0.14:1.52
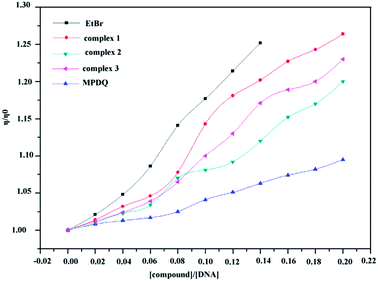 |
| | Fig. 14 Relative viscosity of ct-DNA solution bound with copper(II) complexes and MPDQ with increasing [compound]/[DNA] ratios in the range from 0.02 to 0.20. EtBr as an intercalative indicator was set for comparison. | |
In summary, the DNA binding studies revealed that the binding mode of the copper(II) complexes with ct-DNA was mainly intercalative, which was supported by the results from DNA-melting, competitive binding, CD and viscosity measurements. The copper(II) complexes induced more significant changes in all the spectra of ct-DNA than free MPDQ, which may be the result of the increased planarity of the complexes after MPDQ coordinated to copper(II) ions and the nature of the copper(II) ion. The DNA binding ability of copper(II) complexes to ct-DNA could be correlated to their antitumor activity.
Conclusions
Three new copper(II) complexes of 4,5-methylenedioxy-1-pyridinedihydroisoquinoline were synthesized and fully characterized. These dihydroisoquinoline-copper(II) complexes exhibited enhanced cytotoxicity against tested human tumor cell lines compared to free MPDQ and the corresponding copper(II) salts, suggesting a synergistic effect between MPDQ and copper(II) ions and displayed a selective cytotoxicity to the tested tumor cells. Complex 1 can induce apoptotic death in BEL-7404 cells by triggering S phase cell cycle arrest; moreover, mitochondrial membrane potential and caspase-3 activation assays indicated that the pathway of apoptosis induced by complex 1 correlated with the intrinsic pathway of the mitochondria. The results were further confirmed by analyzing the key proteins of the mitochondrial pathway. ICP-MS testing indicated that complex 1 could enter cells and interact with the DNA of the nuclei. The most probable binding mode of copper(II) complexes with ct-DNA is intercalation, according to the DNA binding assays.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (Nos. 21271051, 21401031, 21431001, 81473102), the National Basic Research Program of China (No. 2012CB723501), the IRT1225, and the Natural Science Foundation of Guangxi Province (Nos.2014GXNSFBA118035) as well as the “BAGUI Scholar” program of Guangxi, China.
Notes and references
- Y. Jung and S. J. Lippard, Chem. Rev., 2007, 107, 1387–1407 CrossRef CAS PubMed.
- J. J. Wilson and S. J. Lippard, J. Med. Chem., 2012, 55, 5326–5336 CrossRef CAS PubMed.
- E. Wexselblatt and D. Gibson, J. Inorg. Biochem., 2012, 117, 220–229 CrossRef CAS PubMed.
- C. P. Tan, S. S. Lai, S. H. Wu, S. Hu, L. J. Zhou, Y. Chen, M. X. Wang, Y. P. Zhu, W. Lian, W. L. Peng, L. N. Ji and A. L. Xu, J. Med. Chem., 2010, 53, 7613–7624 CrossRef CAS PubMed.
- B. M. Zeglis, V. Divilov and J. S. Lewis, J. Med. Chem., 2011, 54, 2391–2398 CrossRef CAS PubMed.
- Z.-F. Chen, Y. Q. Gu, X. Y. Song, Y. C. Liu, Y. Peng and H. Liang, Eur. J. Med. Chem., 2013, 59, 194–202 CrossRef CAS PubMed.
- Z.-F. Chen, Y.-F. Shi, Y.-C. Liu, X. Hong, B. Geng, Y. Peng and H. Liang, Inorg. Chem., 2012, 51, 1998–2009 CrossRef CAS PubMed.
- I. Bratsos, T. Gianferrara, E. Alessio, C. G. Hartinger, M. A. Jakupec and B. K. Keppler, Bioinorg. Med. Chem., 2011, 151–174 CAS.
- M. E. Bravo-Gomez and L. Ruiz-Azuara, New approaches in the treatment of cancer, ed. M. D. C. Mejia Vazquez and S. Navarro, 2010, pp. 139–172 Search PubMed.
- M. F. Primik, S. Goschl, M. A. Jakupec, A. Roller, B. K. Keppler and V. B. Arion, Inorg. Chem., 2010, 49, 11084–11095 CrossRef CAS PubMed.
- H. Thomadaki, A. Karaliota, C. Litos and A. Scorilas, J. Med. Chem., 2008, 51, 3713–3719 CrossRef CAS PubMed.
- C. Duncan and A. R. White, Metallomics, 2012, 4, 127–138 RSC.
- M. Porchia, A. Dolmella, V. Gandin, C. Marzano, M. Pellei, V. Peruzzo, F. Refosco, C. O. Santini and F. Tisato, Eur. J. Med. Chem., 2013, 59, 218–226 CrossRef CAS PubMed.
- M. Connor, A. Kellett, M. M. Cann, G. Rosair, M. M. Namara, O. Howe, B. S. Creaven, S. M. Clean, A. F. A. Kia, D. O'Shea and M. Devereux, J. Med. Chem., 2012, 55, 1957–1968 CrossRef PubMed.
- G. Murtaza, M. K. Rauf, A. Badshah, M. Ebihara, M. Said, M. Gielen, D. Vosd, E. Dilshad and B. Mirza, Eur. J. Med. Chem., 2012, 48, 26–35 CrossRef CAS PubMed.
- S. Tardito, A. Barilli, I. Bassnetti, M. Tegoni, O. Bussolati, R. Franchi-Gazzola, C. Mucchino and L. Marchio, J. Med. Chem., 2012, 55, 10448–10459 CrossRef CAS PubMed.
- D. Palanimuthu, S. V. Shinde, K. Somasundaram and A. G. Samuelson, J. Med. Chem., 2013, 56, 722–730 CrossRef CAS PubMed.
- K. Somdej, K. Kwanjai and L. Ratsami, J. Nat. Prod., 2007, 70, 1536–1538 CrossRef PubMed.
- D. Cappoen, J. Jacobs, T. N. Van, S. Claessens, G. Diels, R. Anthonissen, T. Einarsdottir, M. Fauville, L. Verschaeve, K. Huygen and N. D. Kimpe, Eur. J. Med. Chem., 2012, 48, 57–68 CrossRef CAS PubMed.
- B. Gerhard, Z. G. Liang, B. Tobias, B. Gabi, K. Thomas, B. Holger, B. Reto and M. Virima, Chem.–Eur. J., 2013, 916–923 Search PubMed.
- A. Bermejo, I. Andreu, F. Suvire, S. LeÂonce, D. H. Caignard, P. Renard, A. Pierrea, R. D. Enriz, D. Cortes and N. Cabedo, J. Med. Chem., 2002, 45, 5058–5068 CrossRef CAS PubMed.
- S. A. Kim, Y. Kwon, J. H. Kim, M. T. Muller and I. K. Chung, Biochemistry, 1998, 37, 16316–16324 CrossRef CAS PubMed.
- M. Goldbrunner, G. Loidl, T. Polossek, A. Mannschreck and E. von Angerer, J. Med. Chem., 1997, 40, 3524–3533 CrossRef CAS PubMed.
- W. J. Houlihan, S. H. Cheon, V. A. Parrino, D. A. Handley and D. A. Larson, J. Med. Chem., 1995, 36, 234–240 CrossRef.
- D. Colangelo, A. L. Ghiglia, I. Viano, G. Cavigiolio and D. Osella, BioMetals, 2003, 16, 553–560 CrossRef CAS.
- U. Bierbach, Y. Qu, T. W. Hambley, J. Peroutka, H. L. Nguyen, M. Doedee and N. Farrell, Inorg. Chem., 1999, 38, 3535–3536 CrossRef CAS PubMed.
- E. S. F. Ma, W. D. Bates, A. Edmunds, L. R. Kelland, T. Fojo and N. Farrell, J. Med. Chem., 2005, 48, 5651–5654 CrossRef CAS PubMed.
- F. Nussbaum, B. Miller, S. Wild, C. S. Hilger, S. Schumann, H. Zorbas, W. Beck and W. Steglich, J. Med. Chem., 1999, 42, 3478–3485 CrossRef PubMed.
- Z.-F. Chen, Y.-C. Liu, L.-M. Liu, H.-S. Wang, S.-H. Qin, B.-L. Wang, H.-D. Bian, B. Yang, H.-K. Fun, H.-G. Liu, H. Liang and C. Orvig, Dalton Trans., 2009, 262–272 RSC.
- Y.-C. Liu, Z.-F. Chen, L.-M. Liu, Y. Peng, X. Hong, B. Yang, H.-G. Liu, H. Liang and C. Orvig, Dalton Trans., 2009, 10813–10823 RSC.
- Z.-F. Chen, Y.-C. Liu, Y. Peng, X. Hong, H. H. Wang, M.-M. Zhang and H. Liang, JBIC, J. Biol. Inorg. Chem., 2012, 17, 247–261 CrossRef CAS PubMed.
- R. Al, J. Adnan, C.-Y. Gao, G. E. Martin, F. T. Lin, M. A. Zemaitis and P. L. Schiff, Planta Med., 1998, 64, 681 CrossRef PubMed.
- A. Bermejo, I. Andreu, F. Suvire, S. Leonce, D. H. Caignard, P. Renard, A. Pierre, R. D. Enriz, D. Cortes and N. Cabedo, J. Med. Chem., 2002, 45, 5058–5068 CrossRef CAS PubMed.
- C.-H. Zheng, J. Chen, J. Liu, X.-T. Zhou, N. Liu, D. Shi, J.-J. Huang, J.-G. Lv, J. Zhu and Y.-J. Zhou, Arch. Pharm. Chem. Life Sci., 2012, 345, 454–458 CrossRef CAS PubMed.
- Y. Zhang, J. Feng, C. Liu, L. Zhang, J. Jiao, H. Fang, L. Su, X. Zhang, J. Zhang, M. Li, B. Wang and W. Xu, Bioorg. Med. Chem., 2010, 18, 1761–1772 CrossRef CAS PubMed.
- K.-B. Huang, H.-Y. Mo, J.-H. Wei, Y.-C. Liu, Z.-F. Chen and H. Liang, Eur. J. Med. Chem., 2015, 100, 5213–5222 CrossRef PubMed.
- K.-B. Huang, Z.-F. Chen, Y.-C. Liu, M. Wang, J.-H. Wei, X.-L. Xie, J.-L. Zhang, K. Hu and H. Liang, Eur. J. Med. Chem., 2013, 70, 640–648 CrossRef CAS PubMed.
- G. M. Sheldrick, Phase Annealing in SHELXL-90: Direct methods for larger structures, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1990, 46, 467 CrossRef.
- G. M. Sheldrick, SHELXS-97, Program for X-ray crystal structure Solution, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- K.-B. Huang, Z.-F. Chen, Y.-C. Liu, Z.-Q. Li, J.-H. Wei, M. Wang, G.-H. Zhang and H. Liang, Eur. J. Med. Chem., 2013, 63, 76–84 CrossRef CAS PubMed.
- P. Zhao, J. W. Huang, W. J. Mei, J. He and L. N. Ji, Spectrochim. Acta, Part A, 2010, 75, 1108–1114 CrossRef PubMed.
- M. C. Alley, D. A. Scudiero, A. Monks, M. L. Hursey, M. J. Czerwinski, D. L. Fine, B. J. Abbott, J. G. Mayo, R. H. Shoemaker and M. R. Boyd, Cancer Res., 1988, 48, 589–592 CAS.
- P. U. Maheswari, S. Barends, S. O. Yaman, P. de Hoog, H. Casellas, S. J. Teat, C. Massera, M. Lutz, A. L. Spek, G. P. v. Wezel, P. Gamez and J. Reedijk, Chem.–Eur. J., 2007, 13, 5213–5222 CrossRef CAS PubMed.
- H. Wang, H. Liu, Z. M. Zheng, K. B. Zhang, T. P. Wang, S. S. Sribastav, W. S. Liu and T. Liu, Apoptosis, 2011, 16, 990–1003 CrossRef CAS PubMed.
- Y. Barenholz, J. Kasparkova, V. Brabec and D. Gibson, Angew. Chem., Int. Ed., 2005, 44, 2885–2887 CrossRef PubMed.
- S. Kashanian, M. M. Khodaei and P. Pakravan, Cell Biol., 2010, 29, 639–646 CAS.
- M. Cory, D. D. Mckee, J. Kagan, D. W. Henry and J. A. Miller, J. Am. Chem. Soc., 1985, 107, 2528–2536 CrossRef CAS.
- G. H. Clever, Y. Söltl, H. Burks, W. Spahl and T. Carell, Chem.–Eur. J., 2006, 12, 8708–8718 CrossRef CAS PubMed.
- V. Rajendiran, M. Murali, E. Suresh, M. Palaniandavar, V. S. Periasamy and M. A. Akbarsha, Dalton Trans., 2008, 2157–2170 RSC.
- S. Schäfe, I. R. Ott Gust and W. S. Sheldrick, Eur. J. Inorg. Chem., 2007, 3034 CrossRef PubMed.
- C. Rajput, R. Rutkaite, L. Swanson, I. Haq and J. A. Thomas, Chem.–Eur. J., 2006, 12, 3034–3046 CrossRef PubMed.
- P. G. Baraldi, A. Bovero, F. Fruttarolo, D. Preti, M. A. Tabrizi, M. G. Pavani and R. Romagnoli, Med. Res. Rev., 2004, 24, 475–528 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 923488–923490. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra15789g |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Xiao-Li Xiea and
Hong Liang*ab
a,
Xiao-Li Xiea and
Hong Liang*ab