
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In vivo pharmacokinetics of T2 contrast agents based on iron oxide nanoparticles: optimization of blood circulation times†
Manuel Pernia
Leal
*ab,
Carmen
Muñoz-Hernández
a,
Catherine C.
Berry
b and
María Luisa
García-Martín
*a
aBIONAND, Andalusian Centre for Nanomedicine and Biotechnology, BIONAND (Junta de Andalucía-Universidad de Málaga), Parque Tecnológico de Andalucía, Málaga, Spain. E-mail: mpernia@bionand.es; mlgarcia@bionand.es
bCentre for Cell Engineering, Glasgow University, Joseph Black Building, G12 8QQ, UK
First published on 1st September 2015
Abstract
Magnetic nanoparticles have been extensively investigated for in vivo nanomedical applications in the last decade. PEG coating improves the solubility and stability of the nanoparticles, and provides stealth properties by preventing opsonization and subsequent removal by the reticuloendothelial system. All these effects conferred by the PEG coating are dependent on its molecular weight. Therefore, the selection of the right MW of PEG is a crucial point in the design of new nanomaterials for in vivo applications. The aim of this work lies in the in vivo optimization of the circulation times of small iron oxide nanoparticles by coating them with PEG of different MWs. PEGylated small superparamagnetic iron oxide nanoparticles (PEG-SPIONs), using PEG MWs ranging from 600 to 8000, were synthesized following a ligand exchange methodology, resulting in highly stable and water-soluble nanoparticles. Semi-quantitative and quantitative MRI studies allowed us to track the pharmacokinetics and biodistribution of intravenously injected PEG-SPIONs (HD < 50 nm) in vivo up to one week. Results show that high MW PEGs (6000–8000) lead to nanoparticle aggregation and low MW PEGs (≤1500) are not able to stabilize the 6 nm iron oxide nanoparticles in physiological medium or confer stealth properties, thus leading to rapid recognition by the RES. In contrast, PEG3000-SPIONs show excellent in vivo behavior, they do not aggregate and they exhibit better stealth properties, giving rise to slower liver uptake and longer circulation times. In conclusion, 3000 Da turned out to be the optimal MW for the PEGylation of small nanoparticles (∼6 nm) designed for biomedical applications in which long circulation times together with moderate liver uptake are desirable.
Introduction
Magnetic nanoparticles (MNPs) emerged as promising multifunctional platforms for both drug delivery and imaging based diagnostics.1–5 However, one of the major limitations of these nanomaterials for in vivo applications is their distribution within the body. Most of the nanomaterials injected intravenously are rapidly sequestered by the reticuloendothelial system (RES), thus reducing their efficacy as diagnostic or therapeutic agents.6 Many efforts have been made to enhance blood circulation times and reduce accumulation in the non-desired tissues by modifying the physico-chemical characteristics of the nanomaterial such as size, surface charge and hydrophilic/hydrophobic balance, which are directly involved in the in vivo distribution.7–12 However, there is another important parameter that has to be taken into account when designing nanomaterials for intravenous administration, the plasma protein adsorption.13–15 It is known that neutral and negatively charged nanomaterials reduce the adsorption of plasma protein thus increasing blood circulation times,16 but in some cases the surface charge of the material is not enough to avoid the protein adsorption and the consequent clearance by RES. A polymer layer on the nanomaterial could create a hydrophilic protective coating that inhibits the opsonization process making them stealth.17 The polymer has to be biocompatible and enhance the solubility and stability of the nanomaterial. Among the different biocompatible polymers reported in the literature, polyethylene glycol (PEG) has been the most commonly used.7,9,18–21 PEGylated molecules have been attached directly to the nanomaterials or used as part of the ligand synthesis that will functionalize the corresponding nanomaterial. In both cases, the PEGylated nanomaterials present an improvement in their solubility and stability in aqueous media given by steric repulsion between the PEG molecules. These parameters are also influenced by the molecular weight (MW) of the PEGylated molecule. For instance, some in vivo studies reported that particles coated with high MW PEG (>5000 Da) and hydrodynamic diameters between 50–150 nm presented longer circulation times and a reduction of liver uptake.22–25 However, in vivo information concerning the effect of PEG on the pharmacokinetics and biodistribution of particles with HD sizes below 50 nm is still rare. In this context, we report the synthesis and characterization of a variety of PEGylated (PEG MW = 600–8000 Da) small iron oxide nanoparticles with high stability in different media and conditions. A detailed study of the in vivo pharmacokinetics of different PEGylated nanoparticles has been conducted in mice using MRI. Parametric MRI allowed us to quantify over time the T2 changes induced by these SPIONs on the main organs involved in the excretion process, namely, liver and kidneys. Using these quantitative data we could follow and compare the long-term pharmacokinetics of the different iron oxide nanoparticles and thus determine the effect of PEG MW and hydrodynamic size on their biodistribution and circulation times. The information provided by this paper will allow the readership to better understand the behaviour of functionalized nanoparticles in vivo, which is a crucial aspect that needs to be taken into account in the design of new nanomaterials for biomedical applications.Results and discussion
Characterization of PEG-SPIONs
The stability of the nanoparticles depends on different factors such as pH and ionic strength of the medium. Therefore, the different MW PEG-SPIONs were transferred into physiological conditions of pH 7.2 and NaCl 0.9%, resulting in black clear solutions for all the PEG-SPIONs but the lowest MW, PEG600-SPION, which collapsed due to the lack of stability in the presence of salts. The soluble PEG-SPIONs exhibited hydrodynamic sizes between 17 and 46 nm (Table 1). It was observed that the PEGylation induced an over-increment of the hydrodynamic radius of the SPIONs with the higher molecular weight PEGs (PEG6000- and PEG8000-SPIONs) with respect to the lower ones (PEG1500- and PEG3000-SPIONs). This is likely due to the fact that higher MWs induce aggregation of nanoparticles, with the corresponding increase of the hydrodynamic diameter. This effect was also detected by transmission electron microscopy (TEM). The TEM images of the PEG6000- and PEG8000-SPIONs (see Fig. S1 in the ESI†) showed the presence of these small aggregates of nanoparticles. On the contrary, monodisperse nanoparticles were observed in the TEM images of lower molecular weight PEG (PEG600-, PEG1500- and PEG3000-SPIONs). Nevertheless, all the PEG-SPIONs with diameters ranging from 1500 to 8000 Da, remained stable over months in physiological conditions. Zeta potential of the PEG-SPIONs resulted to be negative charge at pH 7.2 (Table 1). Further characterization, such as FTIR spectroscopy and thermo-gravimetric analysis, were performed for all the nanoparticles. FTIR spectroscopy showed the presence of the PEG ligand on the nanoparticle surface (Fig. S2†), and TGA analysis indicated a percentage of PEGylate ligand around 40% for all PEG-SPIONs (Fig. S3†). Moreover, the dispersant packing density of each nanoparticle was calculated by TGA and TEM (Table 1). A remarkable decreasing tendency of the number of molecules per nm2 with the MW of PEG was observed from 1.99 molecules per nm2, for the lowest MWs, to 0.35 molecules per nm2, for the highest MWs. This tendency could be due to the steric hindrance among the PEG ligands at the nanoparticle surface, and also could explain the partial aggregation in the higher MW PEG SPIONs.| MNPs | Hydrodynamic size (nm) | Packing density (molecules per nm2) | r 2 (mM−1 s−1) 1.5 T | r 2 (mM−1 s−1) 9.4 T | Z potential (meV) |
|---|---|---|---|---|---|
| 600 | 24 ± 4 | 1.99 | 149 ± 30 | 63.7 ± 0.4 | −18.1 ± 3.1 |
| 1500 | 17 ± 1 | 1.31 | 90 ± 1.4 | 113.6 ± 1.1 | −19.8 ± 1.1 |
| 3000 | 18 ± 3 | 0.71 | 103 ± 11 | 151.1 ± 1.7 | −19.3 ± 0.9 |
| 6000 | 44 ± 2 | 0.25 | 190 ± 1.6 | 253.6 ± 5.5 | −14.4 ± 0.2 |
| 8000 | 46 ± 3 | 0.35 | 180 ± 3.2 | 254.2 ± 7.0 | −9.7 ± 0.7 |
In vitro MR relaxivities of PEG-SPIONs
The relaxivity of the PEG-SPIONs were measured at low and high magnetic fields. At clinical magnetic field, 1.5 T, the PEG-SPIONs showed transverse relaxivities, r2, of 90, 103, 190 and 180 mM−1 s−1 for PEG1500-, PEG3000-, PEG6000- and PEG8000-SPIONs, respectively. This trend is in agreement with the hydrodynamic diameter values. Since the inorganic core is the same for all the nanoparticles, it was expected that the transverse relaxivity, r2, would not change appreciably. However, due to the partial aggregation induced by higher MW PEGs, the transverse relaxivities increased considerably. This effect could affect the in vivo results, giving rise to larger T2 contrast with less amount of nanoparticles or, most importantly, inducing a rapid clearance by the RES. The r2 measurements performed at high magnetic field, 9.4 T, showed the same trend found at low magnetic field (114, 151, 253 to 254 mM−1 s−1 for PEG1500-, PEG3000-, PEG6000- and PEG8000-SPIONs, respectively).Stability of PEG-SPIONs
To demonstrate that the GA-PEG-OH ligands are well attached at the nanoparticle surface conferring high stability to the SPIONs,26–28 it was performed an exhaustive study by measuring the hydrodynamic sizes of the PEG-SPIONs in different solvents and at different concentrations, pH and temperatures. Fig. 1 shows the stability results using different solvents: water, PBS buffer and cell growth medium (DMEM + FBS + Medium 199), at pH 7.2 and 37 °C over 24 h, at concentration of 100 μg mL−1 of PEG-SPIONs. All the PEG-SPIONs presented constant hydrodynamic sizes in the different solvents except for the PEG600-SPIONs in PBS buffer. As commented previously, the presence of salts in the PBS buffer reduces the solubility of the nanoparticles leading to nanoparticle collapse. Intriguingly, this effect was not observed in the other solvent, cell growth medium, where the PEG600-SPIONs conserved similar hydrodynamic sizes over 24 h, in spite of having similar salt concentrations. This behavior could be due to the presence of other substances that confer extra stability to the nanoparticles. An increment in the concentration of PEG-SPIONs up to 500 μg mL−1 (Fig. S4†) did not have an effect on the measured sizes, being similar to the ones observed with a concentration of 100 μg mL−1. | ||
| Fig. 1 Hydrodynamic diameters of PEG-SPIONs (100 μg mL−1) vs. time at 37 °C in (A) water at pH 7.2; (B) PBS buffer at pH 7.2 and (C) cell growth medium. | ||
The stability of PEG at the surface of the nanoparticle was also studied with respect to the temperature and the pH. PEG1500-8000 SPIONs presented no change in their hydrodynamic sizes up to 52 °C in PBS buffer. PEG1500- and PEG3000-SPIONs maintained constant sizes even up to 72 °C. In contrast, PEG6000 and PEG8000-SPIONs collapsed over 52 °C probably due to PEG cross-linking between the small aggregates favored by high temperatures (Fig. 2A). As for the pH, all the PEG-SPIONs remained with stable sizes from pH 7.4 to pH 5.3 (Fig. 2B), except for the PEG600-SPIONs, which collapsed rapidly when the pH was lowered to 6.8 (data not shown).
 | ||
| Fig. 2 Stability measurements of PEG-SPIONs (A) vs. temperature in PBS buffer and (B) vs. pH in water. Cytotoxicity of the PEG-SPIONs by the MTT assay of MG-63 cells treated for 24 h (C) and 72 h (D) at different concentrations. | ||
Finally, the cytotoxicity was also evaluated by performing an MTT assay at 24 h and 72 h of incubation (Fig. 2C and D). Both time-lapse studies showed cell metabolic activities above 80% for all the PEG-SPIONs from 0.1 μg mL−1 to 100 μg mL−1 of MNPs.
Summarizing, the stability studies showed the strong attachment of the different MW GA-PEG ligands to the nanoparticle surfaces, and the MTT assays showed no cytotoxicity after 24 h and 72 h of incubation. These results suggest high biocompatibility for the PEG-SPIONs described herein and support their potential use as in vivo contrast agents.
In vivo biodistributions of PEG-SPIONs
The semi-quantitative approach used to follow the pharmacokinetics over the first hour was carried out to study the rapid uptake and excretion processes, which could have not been tracked by using quantitative MRI, due to the low temporal resolution of this type of measurements. Fig. 3 shows representative pharmacokinetics of the PEGylated-SPIONs and gadobutrol (gadolinium(III) 2,2′,2′′-(10-((2R,3S)-1,3,4-trihydroxybutan-2-yl)-1,4,7,10-tetraazacyclododecane-1,4,7-triyl)triacetate), which have been used as a negative control for liver excretion and positive control for renal excretion. It is well known that Gd-complexones are not recognized by the RES and are rapidly eliminated from the blood stream through the urine. Therefore, we included experiments using one of these complexones for comparison with the PEG-SPIONs commented above, which are expected to be excreted through the liver and do not undergo renal filtration.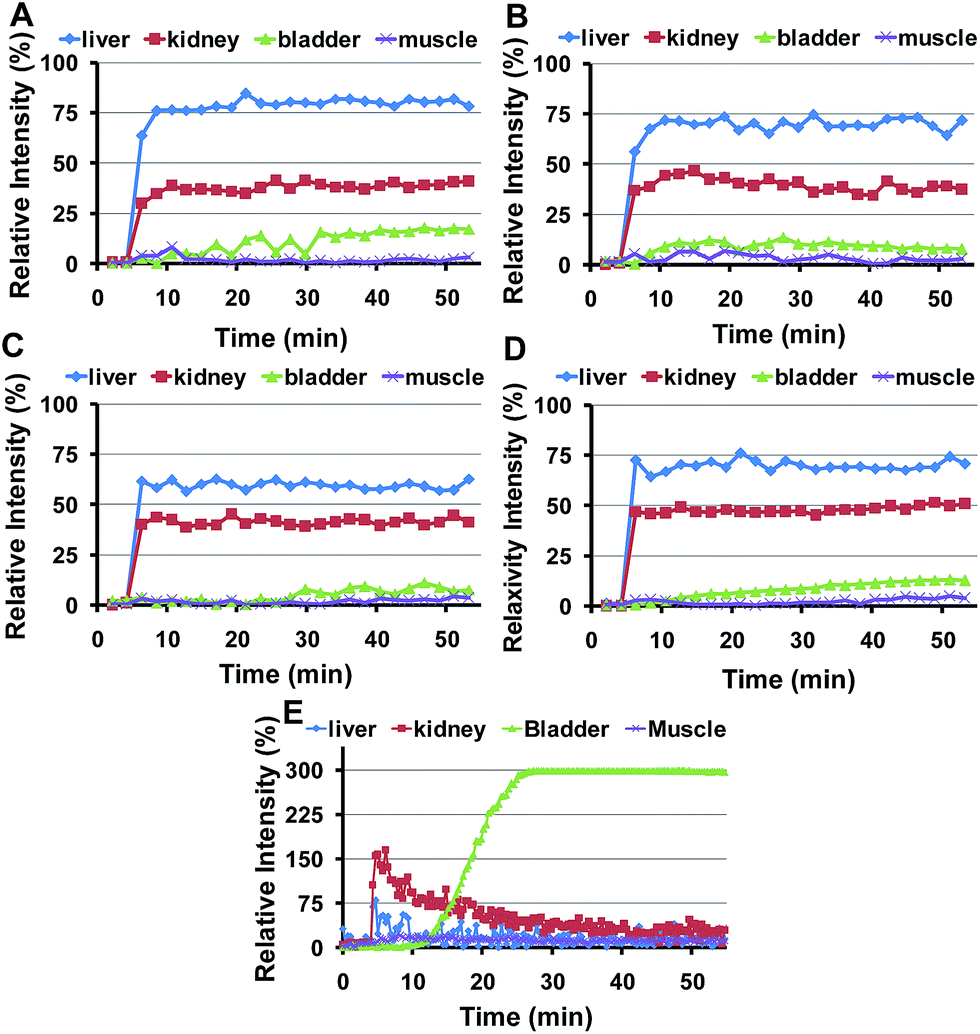 | ||
| Fig. 3 In vivo time courses of PEG-SPIONs after intravenous injection in Balb/c mice. (A) PEG1500-SPIONs; (B) PEG3000-SPIONs; (C) PEG6000-SPIONs; (D) PEG8000-SPIONs; and (E) in vivo time courses of Gadovist® after intravenous injection in Balb/c mice. | ||
Liver uptake
The RE curves (Fig. 3) show that all the PEG-SPIONs are readily taken up by the liver where remain until the end of the experiment. However, there are some important differences in the uptake curves during the first 10 min, with PEG1500- and PEG3000-SPIONs showing slower uptake than PEG6000- and PEG8000-SPIONs. PEG1500-SPIONs reached the maximal concentration approximately 4 min after injection, PEG3000-SPIONs 2 minutes later, and PEG6000- and PEG8000-SPIONs within the first 2 min. This rapid liver uptake of both PEG6000- and PEG8000-SPIONs could be related to the formation of aggregates commented above, which would be immediately recognized by the RES. Interestingly, PEG1500-SPIONs are taken up faster and reach higher concentrations than PEG3000-SPIONs. Since PEG3000-SPIONs have a higher relaxivity, we would expect higher RE values if both nanoparticles reach similar concentrations. However, the RE values are noticeably higher for the PEG1500-SPIONs, indicating that they reach significantly higher concentration in the liver. This behavior suggests that PEG1500-SPIONs have worse stealth properties than PEG3000-SPIONs and hence they are more avidly taken up by the liver.Summing up, we can conclude that high MW PEGs (6000–8000) lead to nanoparticle aggregation, and low MW PEGs (≤1500) are not able to stabilize the SPIONs in physiological medium or confer stealth properties, thus leading to rapid recognition by the RES. In contrast, PEG3000-SPIONs show excellent in vivo behavior, they do not aggregate and exhibit better stealth properties, being the best candidate for biomedical applications in which long circulation times together with moderate liver uptake are desirable.
Long-term pharmacokinetics (Fig. 4–7) showed that only after 1 week the PEGylated SPIONs were fully eliminated from the liver. PEG1500-and PEG8000-SPIONs were eliminated faster, showing around 80% T2 recovery at 48 h, whereas PEG6000-SPIONs showed around 50% T2 recovery and PEG3000-SPIONs only 35% (Fig. 8). This result is in agreement with PEG3000-SPIONs having the longest blood circulation time, followed by PEG6000 and finally PEG1500- and PEG8000-SPIONs.
 | ||
| Fig. 4 Representative T2-weighted MR images of kidneys, liver and muscle at different experimental times after the intravenous injection of PEG-1500 SPIONs. (A and F) T2-weighted images at 0 h; (B and G) at 1 h; (C and H) at 24 h; (D and I) at 48 h; (E and J) at 1 week after injection. Table 2 shows average T2 and ΔT2 values of the different tissues. | ||
 | ||
| Fig. 5 Representative T2-weighted MR images of kidneys, liver and muscle at different experimental times after the intravenous injection of PEG-3000 SPIONs. (A and F) T2-weighted images at 0 h; (B and G) at 1 h; (C and H) at 24 h; (D and I) at 48 h; (E and J) at 1 week after injection. Table 3 shows average T2 and ΔT2 values of the different tissues. | ||
 | ||
| Fig. 6 Representative T2-weighted MR images of kidneys, liver and muscle at different experimental times after the intravenous injection of PEG-6000 SPIONs. (A and F) T2-weighted images at 0 h; (B and G) at 1 h; (C and H) at 24 h; (D and I) at 48 h; (E and J) at 1 week after injection. Table 4 shows average T2 and ΔT2 values of the different tissues. | ||
 | ||
| Fig. 7 Representative T2-weighted MR images of kidneys, liver and muscle at different experimental times after the intravenous injection of PEG-8000 SPIONs. (A and F) T2-weighted images at 0 h; (B and G) at 1 h; (C and H) at 24 h; (D and I) at 48 h; (E and J) at 1 week after injection. Table 5 shows average T2 and ΔT2 values of the different tissues. | ||
 | ||
Fig. 8
T
2 recovery of the (A) liver and (B) kidneys after PEG-SPIONs injections, where  . . | ||
Renal clearance and renal accumulation
Fig. 3E illustrates the renal excretion pharmacokinetics that characterize small molecules, such as Gd-complexones. They reach maximal concentrations in the kidneys very fast and then rapidly decrease as they are being eliminated through the urine. One hour after injection, this type of contrast agents is practically fully cleared out from the bloodstream. On the contrary, the PEGylated SPIONs accumulate in the kidneys (Fig. 3A–D and S5†) because their HDs are well above the size exclusion limit for glomerular filtration, which has been established in 6–8 nm.29 However, as already described by our group,28 the RE increase observed in the bladder demonstrate that some residual renal clearance also occurs. This phenomenon could be explained by shrinkage or partial degradation of the organic shell within the kidneys. In any case, it affects to a very small fraction and therefore it should not have a significant effect on the overall pharmacokinetics of PEGylated-SPIONs.As for renal accumulation, it has been shown that PEGylated NPs are retained in the corpuscles of the kidney cortex in a size-dependent manner,30 with maximal retention for NPs with HD diameters ranging from 76 to 96 nm, and no retention at all for NPs with HD diameters below 25 nm, which could only be found in the peritubular capillaries. According to this study, PEG1500- and PEG3000-SPIONs (HDs 17 and 18 nm, respectively) would not be retained at all, whereas PEG6000- and PEG8000-SPIONs would only show partial retention (HDs 44 nm and 46 nm) (Fig. S5†). Thus, because no retention is expected for the small PEG-SPIONs (PEG1500- and PEG3000-), the signal decay observed in the kidneys can only be attributed to the circulating SPIONs, which shorten the T2 as they pass through the peritubular blood capillaries. In other words, the signal decay observed in the kidneys can be taken as an indicator of the presence of PEG-SPIONs in the bloodstream when the HDs are below 25 nm. According to this, PEG3000-SPIONs were only partially clear out from the bloodstream after one week, showing just 50% T2 recovery in the kidneys (Fig. 8). They were followed by PEG1500-SPIONs, with a 70% kidney T2 recovery. In the case of PEG6000- and PEG8000-SPIONs the interpretation is not so straightforward because part of the remaining signal decay could be attributed to accumulation within the renal corpuscles and not only to circulating nanoparticles.
In summary, PEG3000-SPIONs seem to have the longest circulation times, with significant presence in the bloodstream after one week. Less amount of PEG1500-SPIONs was detected, in agreement with the more efficient elimination through the liver observed for these SPIONs. Finally, the results obtained for PEG6000- and PEG8000-SPIONs are less conclusive because of the complexity underlying the signal T2 decay in the kidneys, which involves both retained and circulating nanoparticles.
Conclusions
This study evidences interesting biodistribution differences depending on the molecular weight and hydrodynamic diameter of PEGylated iron oxide nanoparticles. An exhaustive quantitative and semi-quantitative in vivo analysis was performed on Balb/c mice by magnetic resonance imaging. On the basis of these MRI analyses we demonstrate that PEG3000-SPIONs exhibited the best in vivo behaviour with longer circulation times and slower liver uptake. More in detail, it is observed that PEG6000- and PEG8000-SPIONs reached the maximal nanoparticle concentration in liver and kidneys immediately after injection, whereas the lower MW PEGs reached the maximum after 4 and 6 min (PEG1500- and PEG3000-SPIONs, respectively). Long-term pharmacokinetics showed that only after one week PEGylated SPIONs were fully eliminated from the liver, being PEG3000-SPIONs the ones exhibiting the slower liver excretion, in agreement with PEG3000-SPIONs having the longest circulation time. As expected, PEG-SPIONs exhibited almost no renal excretion and only very little amount was detected in the bladder. On the other hand, the long-term follow up showed that even after one week there was a significant T2 reduction, indicating that the SPIONs were still present at that time. Because nanoparticles are only retained in the corpuscles of the kidney cortex when their HDs exceed 25 nm, the signal decay observed for PEG1500- and PEG3000-SPIONs (HDs of 17 and 18 nm, respectively) can only be attributed to circulating SPIONs, which shorten the T2 as they pass through the peritubular blood capillaries. Therefore, we can conclude that PEG3000-SPIONs were only partially clear out from the bloodstream after one week, showing just 50% T2 recovery in the kidneys, followed by PEG1500-SPIONs, with a 70% kidney T2 recovery. PEG6000-and PEG8000-SPIONs are likely partially retained due to their higher hydrodynamic diameter (HDs 44 nm and 46 nm respectively). In conclusion, these studies show that, for small nanoparticles, with HDs below 50 nm, a PEG coating of 3000 Da confers the best stealth properties, resulting in longer circulation times and slower liver uptake. The information provided by this type of studies can be crucial for the rational design of new nanomaterials intended for in vivo applications, such as molecular imaging or tumour targeting.Experimental
Materials
Reagents were obtained from commercial suppliers and used without further purification. Iron(III) acetylacetonate, oleic acid 99%, oleylamine, benzyl ether, 1,2-hexanedecanediol, gallic acid, poly ethylene glycol 600 Da, 1500 Da, 3000 Da, 6000 Da and 8000 Da, triethylamine, 4-Dimethylaminopyridine, 3-[4,5-dimethylthiazol-2yl]-2,5-diphenyltetrazolium bromide (MTT), phosphate buffered saline (PBS), Dulbecco's Modified Eagle's Medium (DMEM), Medium 199 and Foetal Calf Serum (FCS) were supplied by Sigma Aldrich and Acros Organics. As solvents, Milli-Q water (18.2 MΩ, filtered with filter pore size 0.22 μM) from Millipore, toluene, isopropanol, ethanol, acetone, dimethylsulphoxide (DMSO), hexane, and anhydrous tetrahydrofuran were used (HPLC grade, Acros Organics). Purification of the water-soluble magnetic nanoparticles from free reagents was performed using centrifuge filters PALL with a molecular cut-off of 100 kDa. FTIR spectra were recorded with a FTIR-4100 Jasco.Synthesis of 6 nm iron oxide nanoparticles (SPIONs)
Monodisperse 6 nm Fe3O4 nanoparticles were synthesized following the protocol reported by Sun et al.31 Briefly, 2 mmol of iron acetylacetonate, 10 mmol of hexadecanediol, 6 mmol of dodecylamine, 6 mmol of oleylamine and 20 mL of benzyl ether were mixed and magnetically stirred. The solution was heated to 200 °C for 2 h under a flow of nitrogen, followed by temperature increase to reflux (300 °C) for 1 h. The mixture was cooled down to room temperature by removing the heating mantle. Finally, the sample was washed several times using ethanol, acetone and isopropanol as precipitation agents and centrifugation, followed by re-dispersion in toluene.Synthesis of gallol-derived PEGn-OH ligands (GA-PEGn-OH)
The GA-PEG(n = 600, 1500, 3000, 6000 or 8000 Da)-OH ligands were synthesized following the previously synthetic route reported by Pellegrino and col.32 Briefly, 1 mmol of poly(ethylene glycol) (PEG) (Mw = 600, 1500, 3000, 6000 or 8000 Da), 170 mg of gallic acid (1 mmol) and 23 mg of 4-dimethylaminopyridine (DMAP, 100 μmol) were dissolved in 100 mL of anhydrous THF in a round bottom flask under inert atmosphere. A solution of dicyclohexil carbodiimide (DCC) (1.01 g, 4.9 mmol, 20 mL THF) was added dropwise to the mixture, and it was left stirred overnight at room temperature. The solid urea formed was filtered out and the solvent was removed in the rotaevaporator, yielding a crude GA-PEGn-OH. The crude was dissolved in 100 mL of milli-Q water and adjusted to pH 2. In a separation funnel, the GA-PEGn-OH was extracted from the aqueous phase with diclomethane. The organic phase was concentrated under reduced pressure, yielding the corresponding GA-PEGn-OH. A solution of 0.1 M in chloroform of GA-PEGn-OH ligand was prepared for the ligand exchange procedures.GA-PEG(n = 600, 1500, 3000, 6000 or 8000 Da)-OH ligand exchange on SPIONs
The 6 nm superparamagnetic iron oxide nanoparticles were used as nanoparticle model for the different PEGylated coating. The PEGylation over the SPIONs was performed through a ligand exchange protocol,26,27,32,33 where PEG molecules (600, 1500, 3000 and 8000 Da) are attached covalently to a gallol group to render Gallol-PEG-OH ligands. The different molecular weight ligands were exchanged for the capped surfactants SPIONs thus leading to water soluble 600, 1500, 3000, 6000 and 8000 Da molecular weight PEGylated SPIONs (called from now on PEG600-SPIONs, PEG1500-SPIONs, PEG3000-SPIONs, PEG6000-SPIONs and PEG8000-SPIONs respectively). The excess of the ligand was removed from the PEGylated SPIONs by using the PALL centrifuge filters (MW cut-off: 100 kDa).Transmission electron microscopy (TEM)
TEM images were obtained on a FEI Tecnai G2 Twin microscope operated at an accelerating voltage of 100 kV. TEM samples were prepared by dropping the solution on a carbon-coated copper grid and letting the solvent evaporate.Dynamic light scatering (DLS)
The size distribution and zeta potential measurements of the GA-PEGn-OH were performed on a Zetasizer Nano ZS90 (Malvern, USA).Thermogravimetric analysis (TGA)
Thermogravimetric analysis (TGA) was carried out by using a METTLER TOLEDO model TGA/DSC 1 in the temperature range 30–800 °C with a heating rate of a 10 °C min−1 under N2 flow (50 mL min−1).ICP measurements
Fe concentrations were determined on an Inductively Coupled Plasma High Resolution Mass Spectroscopy (ICP-HRMS, Element XR, Thermo Fisher).In vitro relaxivities
T 2 relaxivities were calculated at two different magnetic fields, 1.5 T (Bruker Minispec) and 9.4 T (Bruker Biospec) using concentrations of GA-PEGn-OH SPIONs between 2.0 and 0.05 mM of Fe in physiological conditions, at 37 °C. T2 was determined using the Carl-Purcell-Meiboom-Gill (CPMG) sequence.Cytotoxicity assays
Briefly, the MG-63 cells were plated at a density of 1 × 104 cells per well in a 96-well plate at 37 °C in 5% CO2 atmosphere (200 μL per well, number of repetitions = 5). After 24 h of culture, the medium in the wells was replaced with fresh medium containing PEG-SPIONs in varying concentrations from 0.1 μg mL−1 to 100 μg mL−1. After 24 h (or 72 h), the medium was removed and 200 μL of fresh medium with MTT (0.5 mg mL−1) was added to each well. After 2 h of incubation at 37 °C and 5% CO2 the medium was removed and the formazan crystals were solubilized with 200 μL of DMSO, and the solution was vigorously mixed to dissolve the reacted dye. The absorbance of each well was read on a microplate reader (Dynatech MR7000 instruments) at 550 nm. The relative cell viability (%) related to control wells containing cell culture medium without nanoparticles was calculated by the equation:
In vivo magnetic resonance imaging (MRI)
In vivo mice experiments were performed in accordance with the ethical guidelines of Andalusian government. Female Balb/c mice (n = 3) with ca. 30 g in weight, provided by Janvier Labs were used. Animals were anesthetized with 1% isoflurane, the tail vein was cannulated and then the animals were placed in the magnet, where respiration and body temperature were monitored throughout the entire MRI experiment. The GA-PEGn-OH SPIONs were administered intravenously via tail vein at a concentration of 5 mg Fe per kg.All the MRI experiments were carried out on a 9.4 T Bruker Biospec system equipped with a 400 mT m−1 gradients and a 40 mm quadrature bird-cage resonator. High resolution T2-weighted images were acquired using a turbo-RARE sequence with respiratory gating (TE = 16 ms, TR = 1000 ms, 4 averages, 156 μm in-plane resolution and 1 mm slice thickness). Quantitative T2 measurements were also performed using a multi-echo spin echo sequence (TEs ranging from 7 ms to 448 ms, TR = 3500 ms, FOV = 4 cm, matrix size = 128 × 128, slice thickness = 1 mm). The time-courses were followed by using a turbo-RARE sequence with the same parameters indicated above, but only 1 average to improve temporal resolution (1 image every 2 minutes). The acquisition scheme was as follows: T2-weighted, quantitative T2, intravenous injection of the GA-PEGn-OH SPIONs, time-course for one hour, quantitative T2 and T2 weighted. The first hour time courses were analyzed semi-quantitatively using the following expression:

Long-term pharmacokinetics were measured by quantitative T2 mapping at 0 h, 1 h, 24 h, 48 h and 1 week. Pharmacokinetics were obtained by calculating the average values within different regions of interest (ROIs) placed on the following tissues: liver, kidney, and muscle. Color-coded RE parametric maps were also calculated on a pixel-by-pixel basis using in-house software written in IDL (RSI, Boulder, USA).
Acknowledgements
Financial support was provided by the Andalusian Ministry of Health (PI2013-0559 to MPL). MPL thanks to the Andalusian Mobility Research Program for Nanomedicine (Fundación Pública Andaluza Progreso y Salud; Andalusian Ministry of Health) and the Talentia Postdoctoral Fellowship Program (grant agreement 267226; Andalusian Knowledge Agency; Andalusian Regional Ministry of Economy, Innovation, Science and Employment) for the Postdoctoral Fellowships. We thank the SCAI-University of Malaga for the TGA and ICP measurements.References
- M. Colombo, S. Carregal-Romero, M. F. Casula, L. Gutiérrez, M. P. Morales, I. B. Böhm, J. T. Heverhagen, D. Prosperi and W. J. Parak, Chem. Soc. Rev., 2012, 41, 4306–4334 RSC.
- A. Figuerola, R. Di Corato, L. Manna and T. Pellegrino, Pharmacol. Res., 2010, 62, 126–143 CrossRef CAS PubMed.
- M. Pernia Leal, A. Torti, A. Riedinger, R. La Fleur, D. Petti, R. Cingolani, R. Bertacco and T. Pellegrino, ACS Nano, 2012, 6, 10535–10545 CAS.
- O. Veiseh, J. W. Gunn and M. Zhang, Adv. Drug Delivery Rev., 2010, 62, 284–304 CrossRef CAS PubMed.
- B. H. Kim, N. Lee, H. Kim, K. An, Y. I. Park, Y. Choi, K. Shin, Y. Lee, S. G. Kwon, H. B. Na, J. G. Park, T. Y. Ahn, Y. W. Kim, W. K. Moon, S. H. Choi and T. Hyeon, J. Am. Chem. Soc., 2011, 133, 12624–12631 CrossRef CAS PubMed.
- B. Wang, X. He, Z. Zhang, Y. Zhao and W. Feng, Acc. Chem. Res., 2013, 46, 761–769 CrossRef CAS PubMed.
- A. S. Karakoti, S. Das, S. Thevuthasan and S. Seal, Angew. Chem., Int. Ed., 2011, 50, 1980–1994 CrossRef CAS PubMed.
- S. Tong, S. Hou, Z. Zheng, J. Zhou and G. Bao, Nano Lett., 2010, 10, 4607–4613 CrossRef CAS PubMed.
- M. D. Howard, M. Jay, T. D. Dziubla and X. Lu, J. Biomed. Nanotechnol., 2008, 4, 133–148 CrossRef CAS.
- S. M. Moghimi, A. C. Hunter and J. C. Murray, Pharmacol. Rev., 2001, 53, 283–318 CAS.
- W. J. Stark, Angew. Chem., Int. Ed., 2011, 50, 1242–1258 CrossRef CAS PubMed.
- J. W. Yoo, E. Chambers and S. Mitragotri, Curr. Pharm. Des., 2010, 16, 2298–2307 CrossRef CAS PubMed.
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS PubMed.
- M. A. Dobrovolskaia and S. E. McNeil, Nat. Nanotechnol., 2007, 2, 469–478 CrossRef CAS PubMed.
- M. A. Dobrovolskaia, P. Aggarwal, J. B. Hall and S. E. McNeil, Mol. Pharm., 2008, 5, 487–495 CrossRef CAS PubMed.
- F. Alexis, E. Pridgen, L. K. Molnar and O. C. Farokhzad, Mol. Pharm., 2008, 5, 505–515 CrossRef CAS PubMed.
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. Vander Elst and R. N. Muller, Chem. Rev., 2008, 108, 2064–2110 CrossRef CAS PubMed.
- M. T. Peracchia, STP Pharma Sci., 2003, 13, 155–161 CAS.
- Y. C. Park, J. B. Smith, T. Pham, R. D. Whitaker, C. A. Sucato, J. A. Hamilton, E. Bartolak-Suki and J. Y. Wong, Colloids Surf., B, 2014, 119, 106–114 CrossRef CAS PubMed.
- J. V. Jokerst, T. Lobovkina, R. N. Zare and S. S. Gambhir, Nanomedicine, 2011, 6, 715–728 CrossRef CAS PubMed.
- J. Milton Harris and R. B. Chess, Nat. Rev. Drug Discovery, 2003, 2, 214–221 CrossRef PubMed.
- R. Gref, Y. Minamitake, M. T. Peracchia, V. Trubetskoy, V. Torchilin and R. Langer, Science, 1994, 263, 1600–1603 CAS.
- C. Fang, B. Shi, Y. Y. Pei, M. H. Hong, J. Wu and H. Z. Chen, Eur. J. Pharm. Sci., 2006, 27, 27–36 CrossRef CAS PubMed.
- T. Maldiney, C. Richard, J. Seguin, N. Wattier, M. Bessodes and D. Scherman, ACS Nano, 2011, 5, 854–862 CrossRef CAS PubMed.
- E. K. U. Larsen, T. Nielsen, T. Wittenborn, L. M. Rydtoft, A. R. Lokanathan, L. Hansen, L. Ostergaard, P. Kingshott, K. A. Howard, F. Besenbacher, N. C. Nielsen and J. Kjems, Nanoscale, 2012, 4, 2352–2361 RSC.
- E. Amstad, S. Zurcher, A. Mashaghi, J. Y. Wong, M. Textor and E. Reimhult, Small, 2009, 5, 1334–1342 CrossRef CAS PubMed.
- A. Riedinger, P. Guardia, A. Curcio, M. A. Garcia, R. Cingolani, L. Manna and T. Pellegrino, Nano Lett., 2013, 13, 2399–2406 CrossRef CAS PubMed.
- M. Pernia Leal, S. Rivera-Fernández, J. M. Franco, D. Pozo, J. M. de La Fuente and M. L. García-Martín, Nanoscale, 2015, 7, 2050–2059 RSC.
- M. Longmire, P. L. Choyke and H. Kobayashi, Nanomedicine, 2008, 3, 703–717 CrossRef CAS PubMed.
- C. H. J. Choi, J. E. Zuckerman, P. Webster and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6656–6661 CrossRef CAS PubMed.
- C. Sun, J. S. H. Lee and M. Q. Zhang, Adv. Drug Delivery Rev., 2008, 60, 1252–1265 CrossRef CAS PubMed.
- A. Riedinger, M. Pernia Leal, S. R. Deka, C. George, I. R. Franchini, A. Falqui, R. Cingolani and T. Pellegrino, Nano Lett., 2011, 11, 3136–3141 CrossRef CAS PubMed.
- E. Amstad, T. Gillich, I. Bilecka, M. Textor and E. Reimhult, Nano Lett., 2009, 9, 4042–4048 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details included TEM images of MNPs, FTIR spectra, thermogravimetric analysis, hydrodynamic size studies, and T2 parametric maps patterns of PEG-SPIONs. See DOI: 10.1039/c5ra15680g |
| This journal is © The Royal Society of Chemistry 2015 |