DOI:
10.1039/C5RA15663G
(Paper)
RSC Adv., 2015,
5, 79041-79049
Two novel cadmium(II) carboxyphosphonates with 3D framework structure: synthesis, crystal structures, luminescence and molecular recognition properties†
Received
5th August 2015
, Accepted 7th September 2015
First published on 7th September 2015
Abstract
Two novel cadmium(II) carboxyphosphonates with 3D framework structure, namely, [Cd3(L)2(H2O)2] (1) and [Cd3Cl2(HL)2(H2O)2] (2) (H3L = H2O3PCH2–NC5H9–COOH) have been synthesized under hydrothermal conditions and structurally characterized. For compound 1, the interconnection of Cd(1)O5N, Cd(2)O6, and CPO3 polyhedra via edge- and corner-sharing forms a 1D chain. The adjacent chains connect with each other by sharing the carboxyphosphonate ligands, thereby generating a 2D layered structure. Neighboring layers are composed in a 3D pillared-layered structure by carboxyphosphonate ligands. Compound 2 exhibits a 3D framework structure. The Cd(1)O4Cl, Cd(2)O4Cl2, and CPO3 polyhedra are interconnected into a 2D layered structure in the bc-plane via corner-sharing, which is further linked to adjacent layers through carboxyphosphonate ligands to form a 3D framework structure. The luminescence properties of compounds 1 and 2 have been investigated. An interesting feature of compound 2 is that it is selective and reversible for sensing of acetone.
Introduction
Recently, metal–organic frameworks (MOFs), which are composed of metal ions or metal clusters and organic ligands, have emerged as a new crystalline porous material with structural diversity and designability.1 Metal organophosphates are an important class of MOF-related materials and have drawn considerable attention not only because of their structural diversity, but also because of their potential applications in such areas as catalysis, photochemistry, magnetism, ion exchanging, materials chemistry, and so on.2 The sum of the distinctive characteristics of the inorganic and organic components and their possible synergistic action could provide novel and intriguing properties for the resulting metal phosphonate materials. Furthermore, the final architecture can be influenced by multifunctional phosphonic acids, solvent molecules, temperature, pH, and coordinated or uncoordinated anions.
A large number of metal phosphonate frameworks with additional functionality have been reported in recent years.3–5 By attaching functional groups such as amine, hydroxyl, and carboxylate groups to the phosphonic acid, our laboratory have designed and synthesized a series of metal phosphonates with framework structures.6 Results from ours and other groups indicate that the carboxyphosphonic acids with additional carboxylic functional groups, such as HOOC–R–PO3H2, HOOC–RN–(CH2PO3H2)2 or HOOC–RNHCH2PO3H2 (R = alkyl or aryl group), have been proved to be very useful ligands for the synthesis of metal phosphonates with framework structures.7 With the aim of exploring novel metal phosphonates with interesting structures and properties, we pay our attention to a multifunctional carboxyphosphonic acid ligand, H2O3PCH2–NC5H9–COOH (H3L), since it can adopt various kinds of coordination modes under different reaction conditions which may result in various interesting structures.8 In this work, by employing carboxyphosphonic acid, H2O3PCH2–NC5H9–COOH (H3L) as the phosphonate ligand, we successfully obtained two novel cadmium(II) carboxyphosphonates with 3D framework structure, namely, [Cd3(L)2(H2O)2] (1) and [Cd3Cl2(HL)2(H2O)2] (2). Herein, we report their synthesis, crystal structures, and luminescence properties. The molecular recognition property of compound 2 has also been studied. To our knowledge, research on the properties of metal phosphonates is mainly focused on the magnetism, luminescence, proton conductivity and ion exchange etc., there are seldom reports about molecular recognition properties of these materials. Molecular recognition, an important detection mean in biological and chemical systems, has been used to investigate straightforward and highly sensitive sensing of small molecules.9 In fact, luminescent MOFs, with high stability and specific structure characteristic, have been testified promising luminescent sensing materials. In recent years, sensing of ions and small molecules utilizing some luminescent MOF materials has been realized and reported.10 Despite great efforts have been made for the sensing applications of the luminescent MOF materials, to the best of our knowledge, no metal phosphonates hybrids have been investigated so far for their molecular recognition properties. To date, only a few investigations on molecular recognition properties of metal phosphonates have recently been reported by our group.11 Further work is in progress to synthesize metal phosphonate hybrids with molecular recognition property.
Experimental
Materials and measurements
The (4-carboxypiperidyl)-N-methylenephosphonic acid, H2O3PCH2–NC5H9–COOH (H3L) was prepared by a Mannich-type reaction according to procedures described previously.12 All other chemicals were obtained from commercial sources and used without further purification. C, H, and N content were determined by using a PE-2400 elemental analyzer. Cd and P content were determined by using an inductively coupled plasma (ICP) atomic absorption spectrometer. IR spectra were recorded on a Bruker AXS TENSOR-27 FT-IR spectrometer with KBr pellets in the range 4000–400 cm−1. FIR spectra were recorded on a NEXUS EURO GC/FT-Infrared Spectrometer with PTFE pellets in the range 400–100 cm−1. The X-ray powder diffraction data was collected on a Bruker AXS D8 Advance diffractometer using Cu-Kα radiation (λ = 1.5418 Å) in the 2θ range of 5–60° with a step size of 0.02° and a scanning rate of 3° min−1. Thermogravimetric (TG) analyses were performed on a Perkin-Elmer Pyris Diamond TG-DTA thermal analyses system in static air with a heating rate of 10 K min−1 from 50 °C to 1100 °C. The luminescence analyses were performed on a HITACHI F-7000 spectrofluorimeter. The luminescent properties of compounds 1 and 2 in the solid state, and compounds 1 and 2 in solvent emulsions were investigated at room temperature. The emulsions were prepared by introducing each sample (1.0 mg) as a powder into different solvents (each 5.0 mL). The luminescence spectra of the emulsions were measured after aging overnight.
Synthesis of [Cd3(L)2(H2O)2] (1). A mixture of CdCl2·2.5H2O (0.11 g, 0.5 mmol) and H3L (0.13 g, 0.5 mmol) was dissolved in 10 mL distilled water. The pH value was adjusted to 7.0 by adding ethylenediamine solution dropwise. The resulting solution was stirred for about 1 h at room temperature, sealed in a 20 mL Teflon-lined stainless steel autoclave, and then heated at 160 °C for 96 h under autogenous pressure. After the mixture was cooled slowly to room temperature, the colorless plate crystals of compound 1 were obtained. Yield: 35.9% (based on Cd). Anal. calcd for C7H13NO6PCd1.50: C, 20.60; H, 3.15; N, 3.50; P, 7.68; Cd, 41.52; found: C, 20.65; H, 3.12; N, 3.53; P, 7.65; Cd, 41.56%. IR (KBr, cm−1): 3444(m), 2928(w), 2874(w), 1618(w), 1543(s), 1440(w), 1363(w), 1294(w), 1118(s), 1039(s), 972(s), 982(m), 863(w), 778(w), 632(w), 571(m), 529(w), 450(w).
Synthesis of [Cd3Cl2(HL)2(H2O)2] (2). A mixture of CdCl2·2.5H2O (0.11 g, 0.5 mmol) and H3L (0.13 g, 0.5 mmol) was dissolved in 10 mL distilled water. The pH value was adjusted to 5.0 by adding ethylenediamine solution dropwise. The resulting solution was stirred for about 1 h at room temperature, sealed in a 20 mL Teflon-lined stainless steel autoclave, and then heated at 160 °C for 96 h under autogenous pressure. After the mixture was cooled slowly to room temperature, the colorless plate crystals of compound 2 were obtained. Yield: 42.6% (based on Cd). Anal. calcd for C14H28Cl2N2O12P2Cd3: C, 18.90; H, 3.25; N, 3.10; P, 6.92; Cl, 8.07; Cd, 38.10; found: C, 18.93; H, 3.22; N, 3.13; P, 6.96; Cl, 8.03; Cd, 38.06%. IR (KBr, cm−1): 3450(s), 2929(s), 2855(m), 1636(m), 1549(m), 1428(m), 1288(w), 1230(w), 1145(m), 1082(s), 996(w), 942(w), 869(w), 772(w), 723(w), 687(w), 587(w), 553(m).
Crystallographic studies
Data collections for compounds 1 and 2 were performed on the Bruker AXS Smart APEX II CCD X-diffractometer equipped with graphite monochromated MoKα radiation (λ = 0.71073 Å) at 293 ± 2 K. An empirical absorption correction was applied using the SADABS program. All structures were solved by direct methods and refined by full-matrix least squares fitting on F2 by SHELXS-2013.13 All non-hydrogen atoms were refined with anisotropic thermal parameters. Hydrogen atoms of organic ligands were generated geometrically with fixed isotropic thermal parameters, and included in the structure factor calculations. Hydrogen atoms for water molecules were not included in the refinement. Details of crystallographic data and structural refinements of compounds 1 and 2 are summarized in Table 1. Selected bond lengths and angles of compounds 1 and 2 are given in Table 2.
Table 1 Crystal data and structure refinements for compounds 1 and 2
| Compounds |
1 |
2 |
| R1 = Σ(|F0| − |FC|)/Σ|F0|. wR2 = [Σw(|F0| − |FC|)2/ΣwF02]1/2. |
| Empirical formula |
C14H26N2O12P2Cd3 |
C14H28Cl2N2O12P2Cd3 |
| Fw |
813.51 |
886.42 |
| Crystal system |
Monoclinic |
Monoclinic |
| Space group |
C2/c |
P2(1)/c |
| a (Å) |
22.4941(15) |
11.6729(6) |
| b (Å) |
11.1377(8) |
8.9390(4) |
| c (Å) |
9.8101(7) |
12.9834(7) |
| β (°) |
103.1000(10) |
110.2660(10) |
| V (Å3) |
2393.8(3) |
1270.87(11) |
| Z |
4 |
2 |
| Dcalcd (mg m−3) |
2.257 |
2.316 |
| μ (mm−1) |
2.835 |
2.884 |
| F(000) |
1576 |
860 |
| Crystal size/mm |
0.33 × 0.06 × 0.05 |
0.35 × 0.30 × 0.24 |
| Theta range (°) |
1.86 to 26.50 |
1.86 to 28.29 |
| Reflections collected/unique |
6652, 2481 (Rint = 0.0155) |
(Rint = 0.0153) |
| GOF on F2 |
1.098 |
1.046 |
| R1, wR2 [I > 2σ (I)]a,b |
0.0270, 0.0833 |
0.0178, 0.0514 |
| R1, wR2 (all data)a,b |
0.0299, 0.0863 |
0.0190, 0.0522 |
Table 2 Selected bond lengths (Å) and angles (°) for compounds 1 and 2a
| Compound 1 |
Compound 2 |
| Symmetry transformations used to generate equivalent atoms. For 1: #1 −x + 1, y, −z + 1/2; #2 −x + 1/2, −y + 1/2, −z; #3 x, −y, z − 1/2; #4 −x + 1, −y, −z + 1; #5 x, −y, z + 1/2 for 2: #1 −x + 1, y + 1/2, −z + 1/2; #2 x − 1, y, z; #3 −x + 2, −y + 1, −z + 1; #4 −x + 1, y − 1/2, −z + 1/2; #5 x + 1, y, z. |
| Distances (Å) |
| Cd(1)–O(3)#1 |
2.222(3) |
Cd(1)–O(1)#1 |
2.1681(14) |
| Cd(1)–O(4)#2 |
2.265(3) |
Cd(1)–O(3)#2 |
2.1818(13) |
| Cd(1)–O(1) |
2.307(3) |
Cd(1)–O(4) |
2.2723(16) |
| Cd(1)–O(2)#3 |
2.310(3) |
Cd(1)–O(5) |
2.4454(16) |
| Cd(1)–N(1) |
2.4209(15) |
Cd(1)–Cl(1)#2 |
2.4912(5) |
| Cd(1)–O(5)#2 |
2.444(4) |
Cd(2)–O(2) |
2.2271(14) |
| Cd(1)–C(7)#2 |
2.6903(15) |
Cd(2)–O(2)#3 |
2.2271(14) |
| Cd(2)–O(1)#1 |
2.291(3) |
Cd(2)–O(6)#3 |
2.2893(16) |
| Cd(2)–O(6)#1 |
2.3077(15) |
Cd(2)–O(6) |
2.2893(16) |
| Cd(2)–O(2)#3 |
2.341(3) |
Cd(2)–Cl(1)#3 |
2.7545(5) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
| Angles (deg) |
| O(3)#1–Cd(1)–O(4)#2 |
148.41(12) |
O(1)#1–Cd(1)–O(3)#2 |
100.09(6) |
| O(3)#1–Cd(1)–O(1) |
88.31(11) |
O(1)#1–Cd(1)–O(4) |
141.60(6) |
| O(4)#2–Cd(1)–O(1) |
123.19(12) |
O(3)#2–Cd(1)–O(4) |
92.69(6) |
| O(3)#1–Cd(1)–O(2)#3 |
90.13(12) |
O(1)#1–Cd(1)–O(5) |
89.95(5) |
| O(4)#2–Cd(1)–O(2)#3 |
93.44(12) |
O(3)#2–Cd(1)–O(5) |
129.32(5) |
| O(1)–Cd(1)–O(2)#3 |
79.19(10) |
O(4)–Cd(1)–O(5) |
55.03(5) |
| O(3)#1–Cd(1)–N(1) |
95.76(10) |
O(1)#1–Cd(1)–Cl(1)#2 |
100.70(4) |
| O(4)#2–Cd(1)–N(1) |
93.76(10) |
O(3)#2–Cd(1)–Cl(1)#2 |
107.74(4) |
| O(1)–Cd(1)–N(1) |
77.41(8) |
O(4)–Cd(1)–Cl(1)#2 |
109.63(6) |
| O(2)#3–Cd(1)–N(1) |
155.66(8) |
O(5)–Cd(1)–Cl(1)#2 |
118.99(4) |
| O(3)#1–Cd(1)–O(5)#2 |
94.23(12) |
O(2)–Cd(2)–O(6)#3 |
90.65(6) |
| O(4)#2–Cd(1)–O(5)#2 |
55.19(12) |
O(2)#3–Cd(2)–O(6)#3 |
89.35(6) |
| O(1)–Cd(1)–O(5)#2 |
170.26(12) |
O(2)–Cd(2)–Cl(1)#3 |
91.34(4) |
| O(2)#3–Cd(1)–O(5)#2 |
110.17(13) |
O(2)#3–Cd(2)–Cl(1)#3 |
88.66(4) |
| N(1)–Cd(1)–O(5)#2 |
92.97(11) |
O(6)#3–Cd(2)–Cl(1)#3 |
87.62(4) |
| O(3)#1–Cd(1)–C(7)#2 |
121.25(9) |
O(6)–Cd(2)–Cl(1)#3 |
92.38(4) |
Results and discussion
Synthesis
Hydrothermal synthesis has been extensively explored for the preparation of metal phosphonates materials. Product composition depends on a number of critical conditions, including pH of the medium, temperature, different metal salts and so on. With the aim to explore optimum methods for obtaining pure phase materials, two systematical experimental investigations have been designed for the system Cd2+/H3L of compounds 1 and 2. The first experiment was designed to investigate the influence of the anions of cadmium salts on the reaction products. Thus, four different cadmium salts (CdCl2·2.5H2O, Cd(Ac)2·2H2O, Cd(NO3)2·4H2O, and CdSO4·8/3H2O) were reacted keeping a constant Cd2+:H3L = 1:1 ratio at their original pH (T = 160 °C, 96 h). Our experiment demonstrates that the final reaction products synthesizing by different cadmium salts exhibit different phases. Cd(NO3)2·4H2O (original pH = 2) and CdSO4·8/3H2O (original pH = 2) acting as reactants synthesize amorphous powders. However, mixture phases (tiny crystals and powder) are obtained by CdCl2·2.5H2O (original pH = 3) and Cd(Ac)2·2H2O (original pH = 4). So we realize that CdCl2·2.5H2O and Cd(Ac)2·2H2O may be more adaptable cadmium salts as the reactant to synthesize compound keeping a constant Cd2+:H3L = 1:1 ratio. Another variable that has a profound impact on the product formation is the pH value of the reaction mixture. To gain a better understanding of the influence of the pH value within the system Cd2+/H3L, a second experiment was designed. The system Cd2+/H3L using two kinds of cadmium salts at different pH, namely, CdCl2·2.5H2O and Cd(Ac)2·2H2O, which have been proved to be more adaptable metal salts as the reactant, was studied under hydrothermal conditions keeping a constant Cd2+:H3L = 1:1 ratio (T = 160 °C, 96 h). The second experiment indicates that mixture phases with Cd(Ac)2·2H2O as cadmium salt are formed at pH = 4, and amorphous powders take place at other pH value under the same synthesis condition. Unexpectedly, two kinds of pure phase of large block single crystals are obtained at pH = 5 and pH = 7 with CdCl2·2.5H2O as cadmium salt. However, the formation of amorphous powders or mixture phases comes into being at other pH value. The powder XRD patterns and the simulated XRD patterns of two title compounds are shown in the ESI (Fig. S1 and S2, ESI†). The powder XRD patterns of compounds 1 and 2 are all essentially in agreement with those simulated from X-ray single-crystal data, confirming that two title compounds are all pure phase. The differences in reflection intensity are probably due to preferred orientation in the powder samples.
Description of the crystal structures
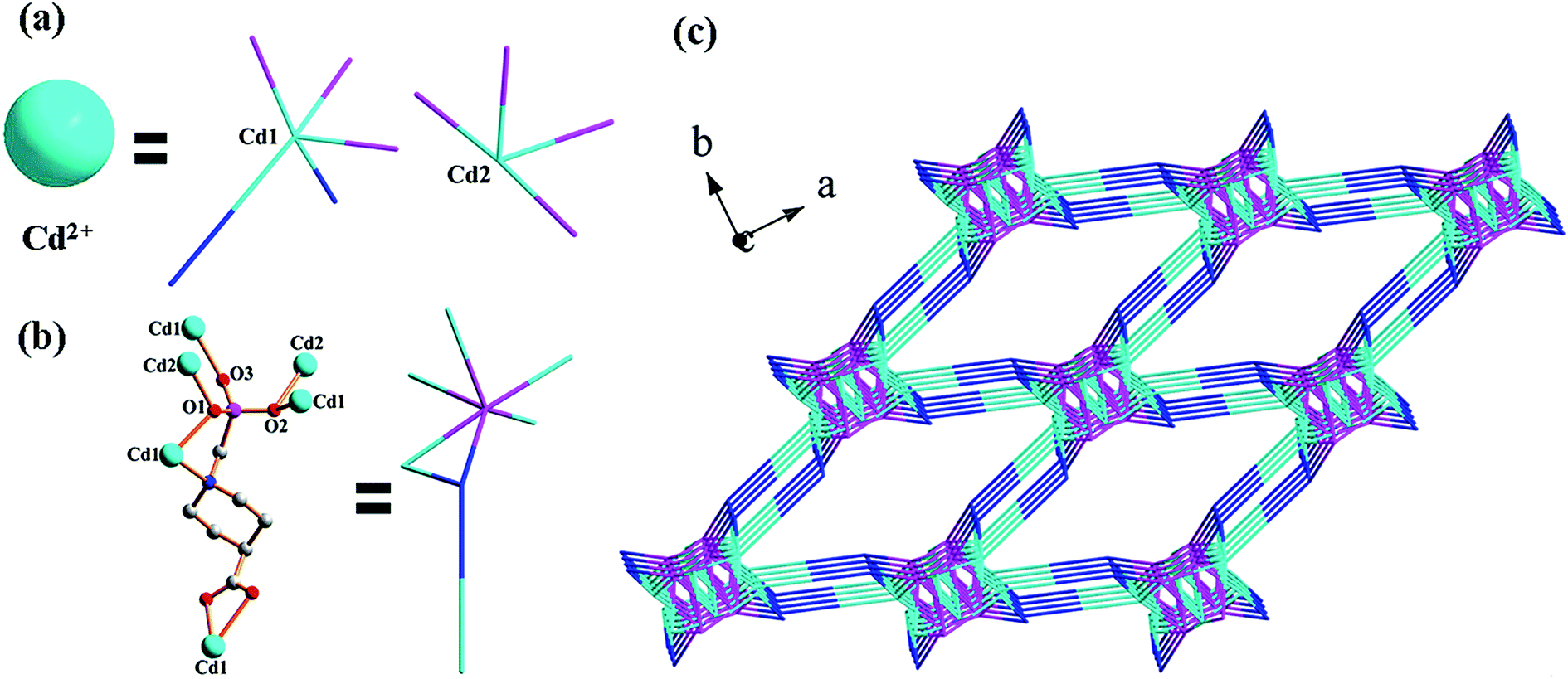
Compound 1 crystallizes in the monoclinic space group C2/c (see Table 1). The asymmetric unit contains two crystallographically unique Cd(II) ions (occupancy: Cd1 100%, Cd2 50%), one L3− anion and one coordinated water molecule.
As shown in Fig. 1, Cd1 ion is six-coordinated by three phosphonate oxygen atoms (O1, O2F, O3B), two carboxylate oxygen atoms (O4C, O5D) and one nitrogen atom (N1) from five separate L3− anions. Cd2 ion is also six-coordinated by four phosphonate oxygen atoms (O1, O1E, O2B, O2F) from four separate L3− anions and two oxygen atoms (O6G, O6) from two coordinated water molecules. The carboxyphosphonate ligand functions as an octadentate metal linkers, it chelates with one Cd1 ion by using one phosphonate oxygen atom (O1) and one nitrogen atom (N1). All oxygen atoms are all involved in the coordination of the metal ions. The phosphonate oxygen atoms (O1, O2) are bidentate metal linkers, whereas the remaining phosphonate oxygen atom (O3) is unidentate (Fig. 2a). The carboxylate group is bidentate, and it chelates with one Cd1 ion by using its two carboxylate oxygen atoms (O4, O5). The Cd–O distances range from 2.222(3) to 2.444(4) and Cd–N distance is 2.4209(15), respectively, which are comparable to those reported for other cadmium(II) carboxyphosphonates previously (see Table 2).14 Based on the charge balance, the carboxyphosphonate ligand is a tervalent anion.
 |
| | Fig. 1 Structure unit of compound 1 showing the atom labeling. Thermal ellipsoids are shown at the 50% probability level. All H atoms are omitted for clarity. Symmetry code for the generated atoms: (A) −x + 1, y, −z + 1/2; (B) −x + 1/2, −y + 1/2, −z; (C) x, −y, z − 1/2; (D) −x + 1, −y, −z + 1; (E) x, −y, z + 1/2. | |
 |
| | Fig. 2 (a) The coordination fashions of H3L in compound 1; (b) the coordination fashions of H3L in compound 2. | |
Compound 1 shows a three-dimensional (3D) framework structure. The Cd(1)O5N, Cd(2)O6 and CPO3 polyhedra are interconnected into a 1D chain along the b-axis via corner- and edge-sharing (Fig. 3a). The adjacent chains connect with each other by sharing the carboxyphosphonate ligands, thereby generating a 2D layered structure in ac-plane (Fig. 3b). Neighboring double layers are bridged by the carboxyphosphonate ligands, leading to a 3D pillared-layered structure (Fig. 4a). The channel system running along the c axis is assembled by 36-atom rings. The size of the channel is estimated to be 6.8 Å (C1–C1) × 8.5 Å (C1–C1) based on structure data. The diameter of the yellow ball inside the channel is 3 Å (Fig. 4b).
 |
| | Fig. 3 (a) The 1D chain structure of compound 1; (b) the 2D layer structure of compound 1. | |
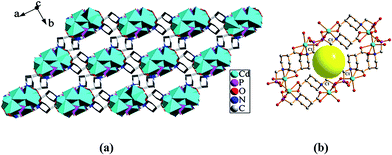 |
| | Fig. 4 (a) The 3D pillared-layered structure of compound 1; (b) the 36-atom channel in compound 1, the diameter of the yellow ball inside the channel is 3 Å. | |
To better understand the structure of compound 1, the topology was analyzed in detail using the TOPOS program.15 Considering the L3− as a 3,6-connected node and Cd2+ as 4-connected and 5-connected node, the total 3-D network exhibits a tetranodal (3,4,5,6)-connected topology with the point symbol of (3·4·5)2(3·44·53·72)2(3·48·52·62·72)2(45·6) (Fig. 5). According to the search results from Reticular Chemistry Structure Resource Database, compound 1 exhibits a completely new topology in MOF chemistry.
 |
| | Fig. 5 Topology of compound 1: (a) the simplified Cd2+ in 4-connected and 5-connected node; (b) the simplified L3− in a 3,6-connected node; (c) the 4-nodal net of compound 1. | |
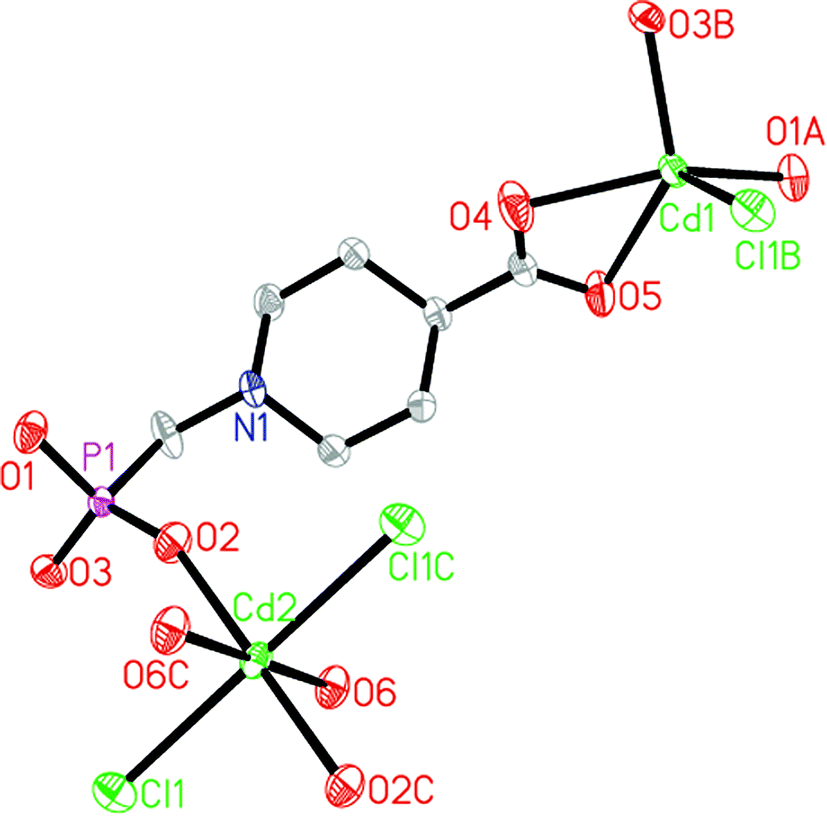
X-ray single crystal diffraction revealed that compound 2 crystallizes in the monoclinic space group P2(1)/c (see Table 1). The asymmetric unit of compound 2 consists of two crystallographically unique Cd(II) ions (occupancy: Cd1 100%, Cd2 50%), one HL2− anion, one chloride anion and one coordinated water molecule. As shown in Fig. 6, Cd1 ion exhibits a five-coordinated environment. Two of the five coordination positions are filled with two phosphonate oxygen atoms (O1A, O3B) from two separate HL2− anions. The remaining sites are occupied by two carboxylate oxygen atoms (O4, O5) from one HL2− anion and one chloride anion (Cl1B). Cd2 ion adopts a six-coordinated environment with two phosphonate oxygen atoms (O2, O2C) from two separate HL2− anions, two oxygen atoms (O6, O6C) from two coordinated water molecules and two chloride anions (Cl1, Cl1C). As shown in Fig. 2b, each HL2− anion acts as a pentadentate ligand, binding four Cd(II) ions through all its phosphonate oxygen atoms and carboxylate oxygen atoms. It chelates one Cd1 ion through two carboxylate oxygen atoms (O4, O5). The three phosphonate oxygen atoms (O1, O2 and O3) are all unidentate. The chloride anion (Cl1) is bidentately bridging between the Cd1 and the Cd2 atoms. The Cd–O and Cd–Cl distances range from 2.1681(14) to 2.4454(16) and from 2.4912(5) to 2.7545(5), respectively, which are in agreement with those reported for other cadmium(II) carboxyphosphonates (see Table 2).16 Based on the requirements of charge balance, the nitrogen atom of each HL2− ligand is protonated.
 |
| | Fig. 6 Structure unit of compound 2 showing the atom labeling. Thermal ellipsoids are shown at the 50% probability level. All H atoms are omitted for clarity. Symmetry code for the generated atoms: (A) −x + 1, y + 1/2, −z + 1/2; (B) x − 1, y, z; (C) −x + 2, −y + 1, −z + 1; (D) −x + 1, y − 1/2, −z + 1/2; (E) x + 1, y, z. | |
Compound 2 exhibits a three-dimensional framework with pillared-layered structure. The Cd(1)O4Cl, Cd(2)O4Cl2 polyhedra and CPO3 tetrahedra are interconnected into a 2D layer structure via corner-sharing (Fig. 7a). Neighboring double layers are bridged by carboxyphosphonate ligands, leading to a 3D pillared-layered structure with channel system running along b-axis (Fig. 7b). The result of connections in this manner is formation of regular channel made up of 24 atoms along the b-axis, which consist of four Cd, two P, four O, two N, and twelve C atoms with the sequences –Cd–Cl–Cd–O–P–C–N–C–C–C–C–O–Cd–Cl–Cd–O–P–C–N–C–C–C–C–O– (Fig. 7c). The size of the channel is estimated to be 12.5 Å (O3–O3) × 4.0 Å (C2–C2) based on structure data. The moderate pore size indicates that such channels can be accessible to a variety of guest molecules, while the chloride atoms and nitrogen atoms on the channel surfaces could be potentially useful for the selective binding of guest molecules.17 From the above results, we conclude that compounds 1 and 2 all show novel three-dimensional framework structure.
 |
| | Fig. 7 (a) The 2D layer structure of compound 2; (b) the 3D pillared-layered structure of compound 2; (c) the 24-atom channel in compound 2. | |
Similarly, the topology about the structure of compound 2 was also analyzed in detail using the TOPOS program. Considering the L3− as a 4-connected node and Cd2+ as 2-connected and 3-connected node, the total 3-D network exhibits a trinodal (2,3,4)-connected topology with the point symbol of (63·82·10)2(63)2(8) (Fig. 8). Compound 2 also exhibits a new topology in MOF chemistry.
 |
| | Fig. 8 Topology of compound 2: (a) the simplified L3− in a 4-connected node; (b) the simplified Cd2+ in 2-connected and 3-connected node; (c) the 3-nodal net of compound 2. | |
Luminescent properties
The luminescent properties of d10 metal complexes have been extensively studied for their potential applications in the field of optical materials. Therefore, the solid state luminescent properties of the free ligand H3L as well as compounds 1 and 2 were investigated at room temperature. The free ligand H3L displays no emission in the visible region. As shown in Fig. 9a, compound 1 exhibits a broad blue fluorescent emission band at λmax = 453 nm upon excitation at 400 nm, which is probably originated from ligand-to-metal charge transfer (LMCT) or metal-to-ligand charge transfer (MLCT).18 Compounds 2 exhibits a emission band between 410 and 450 nm with one strong purple fluorescent emission peak at 417 nm (λex = 370 nm), which is probably due to the metal-to-ligand charge transfer (MLCT) or ligand-to-metal charge transfer (LMCT)18 (Fig. 9b). Unfortunately, the luminescent lifetime of compounds 1 and 2 were not observed, since it is too short to be measured. Under similar measurement conditions, the luminescent behaviors of compounds 1 and 2 are slightly different. The differences in the structures of compounds 1 and 2 result in this phenomenon, due to the luminescence behavior is closely associated with the coordination modes of ligands around the center Cd(II) ions. The further study for Cd(II) luminescent system is underway. The investigation of luminescent properties indicates that compounds 1 and 2 may be good blue-light and purple-light luminescent materials.
 |
| | Fig. 9 (a) The comparison for the solid-state emission spectra of compound 1 at room temperature. (b) The comparison for the solid-state emission spectra of compound 2 at room temperature. | |
Molecular recognition properties
There has been an extensive interest shown in the recognition and sensing of small molecules because of their important roles in biological and chemical systems.19 The differential recognition/binding events with guest molecules confined by the tunable pore sizes and functionalized pore surfaces, which can be transduced into externally optical signals, have enabled the MOFs to become a new type of sensing materials.20 The fluorescence properties of compounds 1 and 2 in different solvent emulsions were investigated for the sensing of small molecules. The emulsions were prepared by introducing 1.00 mg of compounds 1 and 2 powders into 5.00 mL of ethanol, 1-propanol, 1-butanol, 1-pentanol, acetone, butanone, cyclohexanone, DMF and N,N-dimethylacetamide at room temperature.
For compound 1, it did not exhibit the potential for the sensing of guest molecules (Fig. S10, ESI†). While under similar measurement conditions, the fluorescence property indicates that the solvent molecules play an important role in compound 2. Comparing the luminescence intensity of compound 2, cyclohexanone has a minor effect on the luminescence intensity, whereas others exhibit varying degrees of quenching effects. Acetone has the most significant influence on the luminescence intensity (Fig. 10). For further exploration, we examined the molecular recognition properties of compound 2 in detail. Compound 2 was dispersed in cyclohexanone as the standard emulsion, while the solvent content was gradual increased to monitor the emissive response. As show in Fig. 11, while the content of acetone solvent in the emulsion of compound 2 was increased, the fluorescence intensity was evidence quenched. And the fluorescence properties of compound 2 emulsion did not exhibit regular variation in the presence of various contents of 1-butanol, 1-pentanol, 1-propanol, butanone, ethanol, N,N-dimethylacetamide and DMF (Fig. S11–S17, ESI†). Herein, the changes of luminescent intensity on compound 2 were just particular and regular by increasing the content of acetone. It is further indicated that compound 2 showed a selective sieving function to acetone solvent.
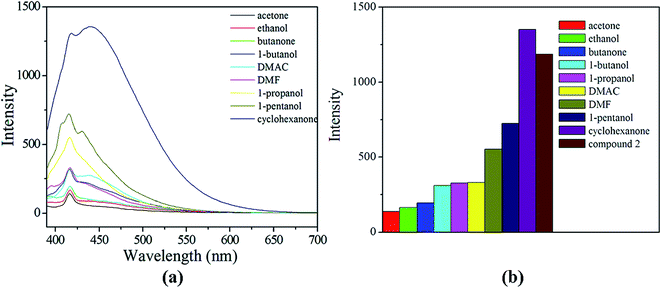 |
| | Fig. 10 (a) The emission spectra and (b) the transition intensities of compound 2 introduces into various pure solvents when excited at 370 nm. | |
 |
| | Fig. 11 The fluorescence properties of compound 2 emulsion in the presence of various contents of acetone solvent (a), respectively (by the frequency of 10 percent every time, from 0 percent to 90 percent). And the luminescence intensities of compound 2 introduced into various contents of acetone solvent (b). | |
The results demonstrate that compound 2 may be used as a promising material for detection of acetone, corresponding to the reported Cd-MOF materials.21,22 The quenching behavior of the acetone might be ascribed to the interaction between “C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O” of acetone and the framework of compound 2. The most significant structural feature of compound 2 is the presence of three-dimensional framework structure within the uncoordinated nitrogen atoms and coordinated chloride atoms, highlighting the potential for its recognition of small molecules. We surmise that the uncoordinated nitrogen atoms and coordinated chloride atoms might enhance the interaction between compound 2 and the small molecules. Although the mechanism of response to organic solvents is still not clear at this moment, the interaction of the crystals and solvent plays an important role in such solvent-dependent luminescent property. The nitrogen atoms are coordinated and there are no chloride atoms in the structures of compounds 1, so the interactions between compounds and solvent molecules are inexistent. Thus, only compound 2 can be a good material used for the sensing of acetone molecules. To check the structural transformation, the PXRD measurement of compound 2 into various solvent after aging has also been done (Fig. S18, ESI†). The powder X-ray diffraction shows that the original framework is not changed. The highly sensing function of compound 2 indicates the promise of this type of luminescent materials for the sensing of substrates in biological systems.23
O” of acetone and the framework of compound 2. The most significant structural feature of compound 2 is the presence of three-dimensional framework structure within the uncoordinated nitrogen atoms and coordinated chloride atoms, highlighting the potential for its recognition of small molecules. We surmise that the uncoordinated nitrogen atoms and coordinated chloride atoms might enhance the interaction between compound 2 and the small molecules. Although the mechanism of response to organic solvents is still not clear at this moment, the interaction of the crystals and solvent plays an important role in such solvent-dependent luminescent property. The nitrogen atoms are coordinated and there are no chloride atoms in the structures of compounds 1, so the interactions between compounds and solvent molecules are inexistent. Thus, only compound 2 can be a good material used for the sensing of acetone molecules. To check the structural transformation, the PXRD measurement of compound 2 into various solvent after aging has also been done (Fig. S18, ESI†). The powder X-ray diffraction shows that the original framework is not changed. The highly sensing function of compound 2 indicates the promise of this type of luminescent materials for the sensing of substrates in biological systems.23
Conclusions
In summary, two novel cadmium(II) carboxyphosphonates with 3D framework structure, namely, [Cd3(L)2(H2O)2] (1) and [Cd3Cl2(HL)2(H2O)2] (2) have been successfully synthesized under hydrothermal conditions. We have systematically investigated the influence of different metal salts and pH value on the synthesis of two title compounds. For compound 1, the interconnection of Cd(1)O5N, Cd(2)O6, and CPO3 polyhedra via edge- and corner-sharing forms a 1D chain, and the adjacent chains connect with each other by sharing the carboxyphosphonate ligands, thereby generating a 3D pillared-layered structure. In compound 2, Cd(1)O4Cl, Cd(2)O4Cl2, and CPO3 polyhedra are interconnected into a 2D layered structure in bc-plane via corner-sharing, which is further linked to adjacent layers through carboxyphosphonate ligands to form a 3D framework structure. Structural simplification of compounds 1 and 2 reveal interesting tetranodal (3,4,5,6)-connected and trinodal (2,3,4)-connected topological nets. The luminescent properties of compounds 1 and 2 indicate that they are good candidates for blue-light and purple-light luminescent materials. Compound 2 exhibits a very high quenching effect with acetone, making it promising for use as a luminescent probe of acetone. This work may enrich the family of luminescent MOFs chemistry and expand the potential applications of such materials.
Acknowledgements
This work is supported by the National Natural Science Foundation of China (Grant No. 21371085).
Notes and references
-
(a) S. R. Batten and R. Robson, Angew. Chem., Int. Ed., 1998, 37, 1460 CrossRef;
(b) O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705 CrossRef CAS PubMed;
(c) D. Bradshaw, S. El-Hankari and L. Lupica-Spagnolo, Chem. Soc. Rev., 2014, 43, 5431 RSC;
(d) V. Stavila, A. A. Talin and M. D. Allendorf, Chem. Soc. Rev., 2014, 43, 5994 RSC.
-
(a) A. Clearfield and K. Demadis, Metal Phosphonate Chemistry: From Synthesis to Applications, Published by the Royal Society of Chemistry, Oxford, 2012, p. 164 Search PubMed;
(b) P. Nugent, Y. Belmabkhout, S. D. Burd, A. J. Cairns, R. Luebke, K. Forrest, T. Pham, S. Ma, B. Space, L. Wojtas, M. Eddaoudi and M. J. Zaworotko, Nature, 2013, 495, 80 CrossRef CAS PubMed;
(c) J. T. Rhule, C. L. Hill and D. A. Judd, Chem. Rev., 1998, 98, 327 CrossRef CAS PubMed;
(d) W. Y. Gao, Y. Chen, Y. Niu, K. Williams, L. Cash, P. J. Perez, L. Wojtas, J. Cai, Y. S. Chen and S. Ma, Angew. Chem., Int. Ed., 2014, 53, 2615 CrossRef CAS PubMed;
(e) Y. Q. Wang, Q. Yue, Y. Qi, K. Wang, Q. Sun and E. Q. Gao, Inorg. Chem., 2013, 52, 4259 CrossRef CAS PubMed.
-
(a) J. M. Rueff, O. Perez, V. Caignaert, G. Hix, M. Berchel, F. Quentel and P. A. Jaffrès, Inorg. Chem., 2015, 54, 2152 CrossRef CAS PubMed;
(b) A. N. Alsobrook, B. G. Hauser, J. T. Hupp, E. V. Alekseev, W. Depmeierc and T. E. Albrecht-Schmitt, Chem. Commun., 2010, 46, 9167 RSC;
(c) D. P. Dong, L. Liu, Z. G. Sun, C. Q. Jiao, Z. M. Liu, C. Li, Y. Y. Zhu, K. Chen and C. L. Wang, Cryst. Growth Des., 2011, 11, 5346 CrossRef CAS.
-
(a) E. Balogh, M. Mato-Iglesias, C. Platas-lglesias, E. Toth, K. Djanashvili, J. A. Peters, A. Beblas and T. Rodriguez-Blas, Inorg. Chem., 2006, 45, 8719 CrossRef CAS PubMed;
(b) X. F. Li, T. F. Liu, Q. P. Lin and R. Cao, Cryst. Growth Des., 2010, 10, 608 CrossRef CAS;
(c) M. Feyand, C. Näther, A. Rothkirch and N. Stock, Inorg. Chem., 2010, 49, 11158 CrossRef CAS PubMed.
-
(a) R. B. Fu, S. M. Hu and X. T. Wu, Cryst. Growth Des., 2014, 14, 6197 CrossRef CAS;
(b) W. Y. Dan, X. F. Liu, M. L. Deng, Y. Ling, Z. X. Chen and Y. M. Zhou, Dalton Trans., 2015, 44, 3794 RSC.
-
(a) Y. Y. Zhu, Z. G. Sun, H. Chen, J. Zhang, Y. Zhao, N. Zhang, L. Liu, X. Lu, W. N. Wang, F. Tong and L. C. Zhang, Cryst. Growth Des., 2009, 9, 3228 CrossRef CAS;
(b) L. Liu, Z. G. Sun, N. Zhang, Y. Y. Zhu, Y. Zhao, X. Lu, F. Tong, W. N. Wang and C. Y. Huang, Cryst. Growth Des., 2010, 10, 406 CrossRef CAS;
(c) K. Chen, Z. G. Sun, Y. Y. Zhu, Z. M. Liu, F. Tong, D. P. Dong, J. Li, C. Q. Jiao, C. Li and C. L. Wang, Cryst. Growth Des., 2011, 11, 4623 CrossRef CAS;
(d) D. P. Dong, Z. G. Sun, F. Tong, Y. Y. Zhu, K. Chen, C. Q. Jiao, C. L. Wang, C. Li and W. N. Wang, CrystEngComm, 2011, 13, 3317 RSC;
(e) F. Tong, Z. G. Sun, K. Chen, Y. Y. Zhu, W. N. Wang, C. Q. Jiao, C. L. Wang and C. Li, Dalton Trans., 2011, 40, 5059 RSC;
(f) W. Chu, Z. G. Sun, C. Q. Jiao, Y. Y. Zhu, S. H. Sun, H. Tian and M. J. Zheng, Dalton Trans., 2013, 42, 8009 RSC.
-
(a) P. F. Wang, D. K. Cao, S. S. Bao, H. J. Jin, Y. Z. Li, T. W. Wang and L. M. Zheng, Dalton Trans., 2011, 40, 1307 RSC;
(b) J. T. Li, D. K. Cao, T. Akutagawa and L. M. Zheng, Dalton Trans., 2010, 39, 8606 RSC;
(c) P. O. Adelani, A. G. Oliver and T. E. Albrecht-Schmitt, Cryst. Growth Des., 2011, 11, 1966 CrossRef CAS;
(d) N. G. Armatas, D. G. Allis, A. Prosvirin, G. Carnutu, C. J. O'Connor, K. Dunbar and J. Zubieta, Inorg. Chem., 2008, 47, 832 CrossRef CAS PubMed;
(e) K. Barthelet, M. Nogues, D. Riou and G. Férey, Chem. Mater., 2002, 14, 4910 CrossRef CAS;
(f) H. Tan, W. Chen, D. Liu, Y. Li and E. Wang, Dalton Trans., 2010, 39, 1245 RSC.
-
(a) S. Bauer, T. Bein and N. Stock, Inorg. Chem., 2005, 44, 5882 CrossRef CAS PubMed;
(b) J. L. Song and J. G. Mao, J. Mol. Struct., 2005, 740, 181 CrossRef CAS PubMed;
(c) N. Stock and T. Bein, Angew. Chem., Int. Ed., 2004, 43, 749 CrossRef CAS PubMed;
(d) S. F. Tang, J. L. Song and J. G. Mao, Eur. J. Inorg. Chem., 2006, 2011 CrossRef CAS PubMed.
-
(a) M. Zhang, Z. J. Pu, X. L. Chen, X. L. Gong, A. X. Zhu and L. M. Yuan, Chem. Commun., 2013, 49, 5201 RSC;
(b) B. L. Chen, S. C. Xiang and G. D. Qian, Acc. Chem. Res., 2010, 43, 1115 CrossRef CAS PubMed;
(c) B. L. Chen, L. B. Wang, F. Zapata, G. D. Qian and E. B. Lobkovsky, J. Am. Chem. Soc., 2008, 130, 6718 CrossRef CAS PubMed.
-
(a) H. Xu, F. Liu, Y. J. Cui, B. L. Chen and G. D. Qian, Chem. Commun., 2011, 47, 3153 RSC;
(b) J. Y. Guo, H. Xu, S. Q. Su, J. F. Cai, S. Dang, S. C. Xiang, G. D. Qian, H. J. Zhang, M. O'Keeffe and B. L. Chen, Chem. Commun., 2011, 47, 5551 RSC;
(c) Y. Q. Xiao, Y.
J. Cui, Q. Zheng, S. C. Xiang, G. D. Qian and B. L. Chen, Chem. Commun., 2010, 46, 5503 RSC;
(d) Z. J. Zhang, S. C. Xiang, X. T. Rao, Q. Zheng, F. R. Fronczek, G. D. Qian and B. L. Chen, Chem. Commun., 2010, 46, 7205 RSC.
-
(a) S. P. Shi, Y. Y. Zhu, Z. G. Sun, W. Zhou, L. L. Dai, M. X. Ma, W. Z. Li, H. Luo and T. Sun, Cryst. Growth Des., 2014, 14, 1580 CrossRef CAS;
(b) W. Zhou, Y. Y. Zhu, C. Q Jiao, Z. G. Sun, S. P. Shi, L. L. Dai, T. Sun, W. Z. Li, M. X. Ma and H. Luo, CrystEngComm, 2014, 16, 1174 RSC;
(c) L. L. Dai, Y. Y. Zhu, C. Q. Jiao, Z. G. Sun, S. P. Shi, W. Zhou, W. Z. Li, T. Sun, H. Luo and M. X. Ma, CrystEngComm, 2014, 16, 5050 RSC;
(d) T. Sun, C. Q. Jiao, W. Z. Li, Z. G. Sun, C. Ma, Y. Y. Zhu, M. X. Ma, H. Luo, X. W. Zhang and M. L. Wang, RSC Adv., 2015, 5, 26410 RSC.
- C. Serre, N. Stock, T. Bein and G. Férey, Inorg. Chem., 2004, 43, 3159 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
-
(a) J. L. Song, J. G. Mao, Y. Q. Sun, H. Y. Zeng, R. K. Kremer and A. Clearfield, Solid State Chem., 2004, 177, 633 CrossRef CAS PubMed;
(b) Z. M. Sun, J. G. Mao, Y. Q. Sun, H. Y. Zenga and A. Clearfield, New J. Chem., 2003, 27, 1326 RSC.
- W. Y. Dan, X. F. Liu, M. L. Deng, Y. Ling, Z. X. Chen and Y. M. Zhou, Dalton Trans., 2015, 44, 3794 RSC.
-
(a) N. Stock, Solid State Chem., 2002, 4, 1089 CAS;
(b) J. G. Mao, Z. Wang and A. Clearfield, Inorg. Chem., 2002, 41, 6106 CrossRef CAS PubMed.
- Z. Y. Guo, X. Z. Song, H. P. Lei, H. L. Wang, S. Q. Su, H. Xu, G. D. Qian, H. J. Zhang and B. L. Chen, Chem. Commun., 2015, 51, 376 RSC.
- K. R. Ma, J. N. Xu, L. R. Zhang, J. Shi, D. J. Zhang, Y. L. Zhu, Y. Fan and T. Y. Song, New J. Chem., 2009, 33, 886 RSC.
-
(a) T. M. Reineke, M. Eddaoudi, M. Fehr, D. Kelley and O. M. Yaghi, J. Am. Chem. Soc., 1999, 121, 1651 CrossRef CAS;
(b) O. M. Yaghi, G. Li and H. Li, Nature, 1995, 378, 703 CrossRef CAS PubMed;
(c) H. Li, M. Eddaoudi, T. L. Groy and O. M. Yaghi, J. Am. Chem. Soc., 1998, 120, 8571 CrossRef CAS.
-
(a) Z. Y. Guo, H. Xu, S. Q. Su, J. F. Cai, S. Dang, S. C. Xiang, G. D. Qian, H. J. Zhang, M. O'Keeffe and B. L. Chen, Chem. Commun., 2011, 47, 5551 RSC;
(b) Y. A. Li, S. K. Ren, Q. K. Liu, J. P. Ma, X. Y. Chen, H. M. Zhu and Y. B. Dong, Inorg. Chem., 2012, 51, 9629 CrossRef CAS PubMed.
- F. Y. Yi, W. T. Yang and Z. M. Sun, J. Mater. Chem., 2012, 22, 23201 RSC.
- J. M. Zhou, W. Shi, H. M. Li, H. Li and P. Cheng, J. Phys. Chem. C, 2013, 118, 416 Search PubMed.
-
(a) B. L. Chen, Y. Yang, F. Zapata, G. N. Lin, G. D. Qian and E. B. Lobkovsky, Adv. Mater., 2007, 19, 1694 Search PubMed;
(b) H. Xu, F. Liu, Y. J. Cui, B. L. Chen and G. D. Qian, Chem. Commun., 2011, 47, 3153 RSC;
(c) Y. J. Cui, Y. F. Yue, G. D. Qian and B. L. Chen, Chem. Rev., 2012, 112, 1126 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: XRD patterns, IR and FIR spectra, IR and FIR discussion, thermal analyses and XRD patterns of the final products in the thermal decomposition for compounds 1 and 2. CCDC 1413756 and 1413757. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra15663g |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.