
Electrochemistry of nonplanar copper(II) tetrabutano- and tetrabenzotetraarylporphyrins in nonaqueous media
Lina Yeab,
Zhongping Ou*a,
Yuanyuan Fangac,
Songlin Xuea,
Yang Songc,
Liping Wanga,
Mengli Wanga and
Karl M. Kadish*c
aSchool of Chemistry and Chemical Engineering, Jiangsu University, Zhenjiang 212013, P. R. China. E-mail: zpou2003@yahoo.com
bCollege of Computer, Jilin Normal University, Siping 136000, P. R. China
cDepartment of Chemistry, University of Houston, Houston, Texas 77204-5003, USA. E-mail: kkadish@uh.edu
First published on 4th September 2015
Abstract
Two series of copper tetraarylporphyrins containing β,β′-fused tetrabutano or tetrabenzo groups were synthesized and characterized as to their electrochemistry and spectroelectrochemistry in nonaqueous media. The examined compounds are represented as butano-(TpYPP)CuII and benzo-(TpYPP)CuII, where TpYPP is the porphyrin dianion and Y is a CH3, H or Cl substituent on the para-position of the four meso-phenyl rings of the compound. Each neutral porphyrin in the two series is ESR active and shows a typical d9 Cu(II) signal in frozen CH2Cl2 solution. Each Cu(II) porphyrin also undergoes two reversible one-electron reductions and two reversible one-electron oxidations in DMF or CH2Cl2 containing 0.1 M tetra-n-butylammonium perchlorate to give a π-anion radical and dianion upon reduction and a π-cation radical and dication upon oxidation. A third one-electron oxidation is also observed for butano-(TpYPP)Cu (Y = CH3 and H) and benzo-(TPP)Cu in PhCN and this process is assigned to the CuII/CuIII transition. The reversible half-wave potential for the first oxidation of each compound in both series is shifted negatively by about 500 mV as compared to E1/2 values for oxidation of the related copper tetraarylporphyrin without the four fused benzo or butano rings while smaller positive shifts of 60 and 300 mV are seen for reduction of the tetrabenzotetraarylporphyrins and tetrabutaotetraarylporphyrins, respectively, as compared to the same redox reactions of the related tetraarylporphyrins. The electrochemically measured HOMO–LUMO gap averages 1.76 ± 0.05 V for benzo-(TpYPP)CuII, 2.04 ± 0.06 V for butano-(TpYPP)CuII and 2.33 ± 0.03 for (TpYPP)Cu in CH2Cl2.
Introduction
It is known that the addition of electron-donating or electron-withdrawing substituents on the meso- and/or β-pyrrole positions of a porphyrin macrocycle can significantly affect the spectra and electrochemical properties of the compound.1,2 The electrochemistry of porphyrins with redox inactive central metals is characterized by two reductions and two oxidations in nonaqueous media, all of which are localized on the macrocyclic π-ring system.1,2 Additional metal-centered reactions may also be observed for porphyrins with transition metal ions, the most studied of which have been the MII/MIII processes involving porphyrins with Fe, Co, Ni or Mn centers.1–6 Porphyrins with AgII and AuII centers can also be oxidized to their +3 oxidation state7–11 but until recently12 it was believed that a similar reaction would not occur for CuII porphyrins which were thought to have a redox inactive central metal ion.1,2A number of meso and/or β-substituted copper porphyrins have been synthesized and characterized as to their spectroscopic properties and electrochemistry1,2,12,13 and these synthetic copper porphyrins have been the subject of numerous investigations.1,2,13–29 Our recent report of a CuII/CuIII porphyrin oxidation involved highly substituted nonplanar compounds12 and we wished to know how changing the planarity or π-ring system of the porphyrin macrocycle by addition of four β,β′-fused butano or benzo groups might affect the redox potentials and electron transfer mechanisms of these copper(II) porphyrins.
Tetrazenzoporphyrins have been recognized for their use in the areas of catalysis, medicine and material science.30 In the present work, two series of copper tetraarylporphyrins containing four fused β,β′-butano or β,β′-benzo rings were synthesized and characterized as to their electrochemistry and spectroelectrochemistry in nonaqueous media. The examined compounds are represented as butano-(TpYPP)CuII and benzo-(TpYPP)CuII, where TpYPP is a dianion of the porphyrin and Y is a CH3, H or Cl substitutent on the para-position of the four meso-phenyl rings. Structures of the examined butanoporphyrins 1b–3b and benzoporphyrins 1c–3c are shown in Chart 1 which also includes the (TpYPP)CuII comparison compounds 1a–3a which are planar and octaethyltetraphenylporphyrin, (OETPP)Cu, and dodecaphenylporphyrin, (DPP)Cu, which are not. Each neutral copper(II) porphyrin in the three series was characterized by electrochemistry, UV-visible spectroscopy, mass spectrometry and ESR spectroscopy. Each reduced and oxidized form of the neutral porphyrin was also characterized by thin-layer spectroelectrochemistry. The effect of the four fused butano or benzo groups on the spectra and electrochemical properties of the compounds is discussed.
 | ||
| Chart 1 Structures of examined copper porphyrins. | ||
Results and discussion
Synthesis and UV-Vis spectra
The synthetic routes to obtain butano-(TpYPP)Cu 1b–3b and benzo-(TpYPP)Cu 1c–3c are shown in Scheme 1. Cyclohexene was transformed into nitrocyclohexene which was reacted with ethyl isocyanoacetate to give a tetrahydroisoindole ester. 4,5,6,7-Tetrahydroisoindole was obtained from the tetrahydroisoindole ester by refluxing with excess KOH and ethylene glycol (EG) for 1.5 h. | ||
| Scheme 1 Synthetic routes for tetrabutanotetraarylporphyrins 1b–3b and tetrabenzotetraarylporphyrins 1c–3c. | ||
The free-base tetrabutanotetraarylporphyrins were synthesized from tetrahydroisoindole and arylaldehyde using a method reported by Beletskaya, Vinogradov and coworkers.31 The Cu(II) butanoporphyrins 1b–3b were then obtained via a reaction between butano-(TpYPP)H2 and Cu(OAc)2 in a mixed solvent of CHCl3 and CH3OH (v/v = 4/1) with a yield of 80–85%. After purification by column chromatography, the copper tetrabutanotetraarylporphyrins were oxidized by 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) in tetrahydrofuran (THF) to give the corresponding copper tetrabenzotetraaryl-porphyrins 1c–3c with the yield ranging from 48 to 55%.
UV-Vis spectra of 1a–3a, 1b–3b and 1c–3c in CH2Cl2 are illustrated in Fig. 1. The (TpYPP)CuII compounds 1a–3a (Fig. 1a) are characterized by a sharp Soret band at 415–418 nm and a low intensity Q band at ∼540 nm while the tetrabutanoporphyrins 1b–3b exhibit red-shifted Soret and Q bands as compared to the (TpYPP)CuII derivatives with the same meso-substituents. In addition, the single Q band of the (TpYPP)CuII complexes is replaced by two Q bands for 1b–3b as seen in the figure. Spectra of the butano-(TpYPP)CuII compounds are similar to the spectrum of (OETPP)Cu which has a Soret band at 430 nm and two Q bands at 568 and 598 nm.32
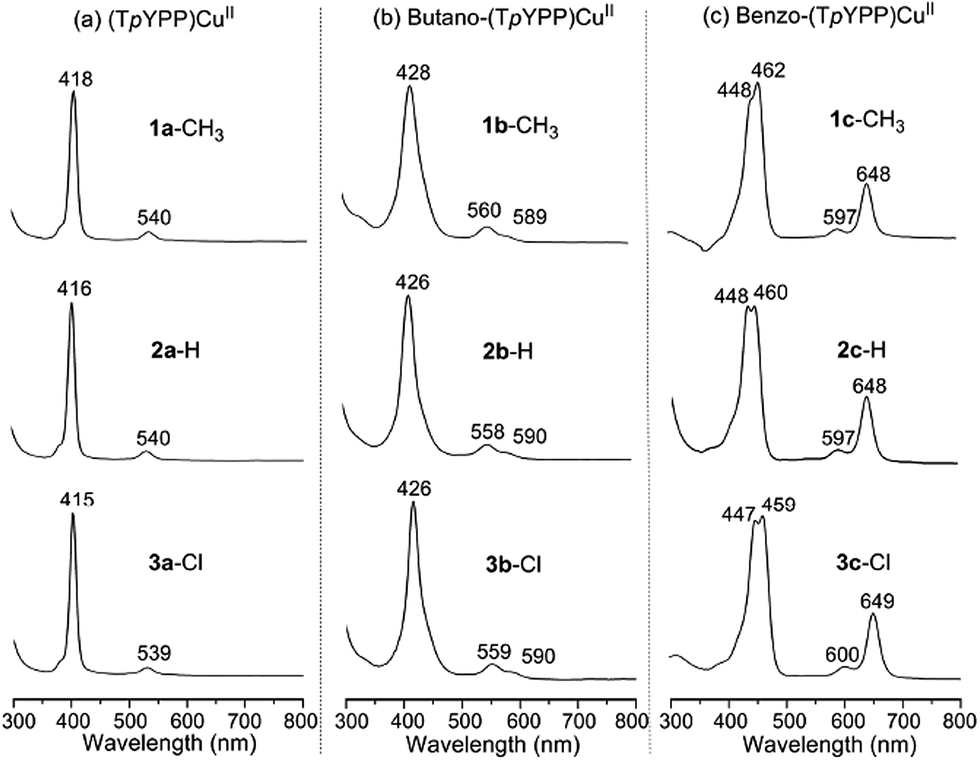 | ||
| Fig. 1 UV/Vis spectra of (a) (TpYPP)CuII 1a–3a, (b) butano-(TpYPP)CuII 1b–3b and (c) benzo-(TpYPP)CuII 1c–3c in CH2Cl2. | ||
A significant difference is seen in UV-Vis spectra of the benzo-(TpYPP)CuII compounds 1c–3c. These porphyrins, which possess four β,β′-fused ring systems, are characterized in CH2Cl2 by a split Soret band at 447–462 nm, an intense Q band at ∼648 nm and a weaker Q band at 597–600 nm (Fig. 1c). The average red shift in the Soret band is about 38 nm as compared to the corresponding tetraarylporphyrins with the same meso-substituents while the highest intensity Q band of benzo-(TpYPP)CuII is shifted by ∼110 nm as compared to (TpYPP)CuII. The longest wavelength Q band for the compounds in Fig. 1c has a relatively high intensity due to the increased conjugation between the four fused benzo groups and the macrocycle of the compounds. This result is consistent with what has been reported for benzoporphyrins having different central metal ions.33–38
ESR characterization
ESR spectra for the tetrabutano and tetrabenzo derivatives of Cu(II) at 110 K in CH2Cl2 are illustrated in Fig. 2 while the ESR parameters are summarized in Table 1. The spectra of butano-(TpYPP)Cu 1b–3b exhibit features typical of monomeric Cu(II) tetrapyrroles,39–41 with g// values ranging from 2.193 to 2.204 (A// = 206–210) and g⊥ ranging from 2.053 to 2.054 (A⊥ = 117–130). No nitrogen superhyperfine is seen in the spectra of Fig. 2a, indicating the lack of a strong interaction between the Cu(II) ion and the four nitrogen atoms under the given experimental conditions.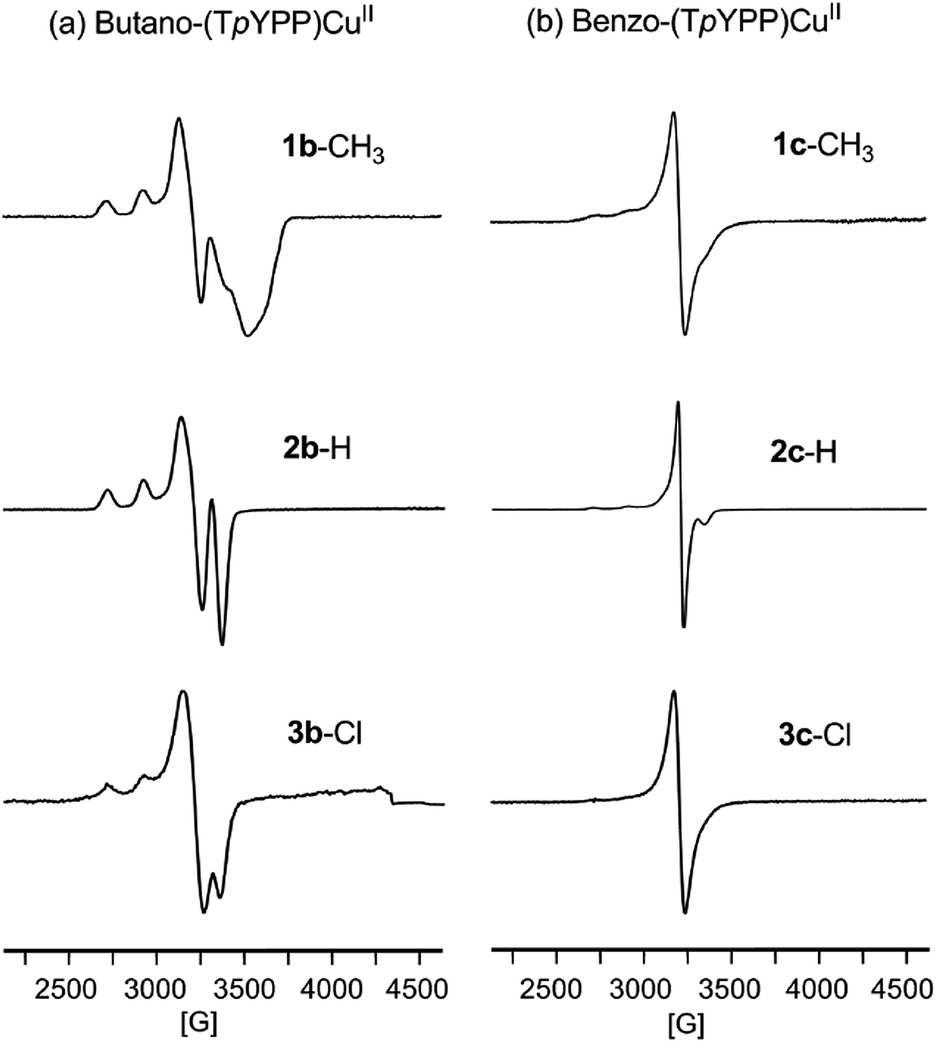 | ||
| Fig. 2 ESR spectra of copper tetrabutanotetraarylporphyrins 1b–3b (∼10−3 M) and tetrabenzotetraarylporphyrins 1c–3c (∼10−3 M) in frozen dichloromethane at 110 K. | ||
| compound | g// | A// | g⊥ | A⊥ |
|---|---|---|---|---|
| (TPP)CuII 2a | 2.219 | 222 | 2.057 | 218 |
| Butano-(TpCH3PP)Cu 1b | 2.200 | 210 | 2.054 | 130 |
| Butano-(TPP)Cu 2b | 2.193 | 206 | 2.054 | 117 |
| Butano-(TpClPP)Cu 3b | 2.204 | 206 | 2.053 | 117 |
| Benzo-(TpCH3PP)Cu 1c | 2.051 | 66 | ||
| Benzo-(TPP)Cu 2c | 2.053 | 41 | ||
| Benzo-(TpClPP)Cu 3c | 2.052 | 66 |
The ESR spectrum of each Cu(II) tetrabenzotetraaryl-porphyrin is characterized by a prominent g⊥ value ranging from 2.051 to 2.053 (A⊥ = 41–66 as seen in Table 1), values consistent what was previously reported for other Cu(II) porphyrins.42–44 It should be pointed out that the Cu(II) hyperfine is ill-defined in spectra of the benzo-(TpYPP)Cu derivatives at 110 K in CH2Cl2 (Fig. 2b) and this can be accounted for by aggregation of these porphyrins under the given experimental conditions.39,45–47
Electrochemistry
The electrochemistry of butano-(TpYPP)Cu 1b–3b and benzo-(TpYPP)Cu 1c–3c was carried out in CH2Cl2, DMF and PhCN containing 0.1 M TBAP. Examples of cyclic voltammograms in CH2Cl2 are shown in Fig. 3 and a summary of the measured reduction and oxidation potentials for each porphyrin in the three solvents is given in Table 2, which includes data for several structurally related Cu(II) porphyrins measured under the same solution conditions. A discussion of the data is given below, first for oxidation and then for reduction, each of which is affected differently by the type of β,β′-fused groups. | ||
| Fig. 3 Cyclic voltammograms of (a) (TPP)CuII 2a, (b) benzo-(TPP)CuII 2c and (c) butano-(TPP)CuII 2b in CH2Cl2 containing 0.1 M TBAP. | ||
| solvent | Compound | Oxidation | Reduction | ΔEd (V) | Ref. | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| 3rd | 2nd | 1st | ΔE(2o-1o) | 1st | 2nd | ΔE(1r−2r) | ||||
| a Irreversible peak potential at a scan rate of 0.1 V s−1.b The solubility of cpds 1c, 3b and 3c is very low and meaningful data could not be obtained in PhCN.c The solubility of cpd 1c is very low in DMF and no redox processes could be observed.d The potential difference between the first oxidation and first reduction (HOMO–LUMO gap). | ||||||||||
| CH2Cl2 | (TpCH3PP)Cu 1a | 1.23 | 0.93 | 0.30 | −1.37 | −1.83a | 0.46 | 2.30 | 48 | |
| (TPP)Cu 2a | 1.30 | 1.04 | 0.26 | −1.28 | −1.71 | 0.43 | 2.32 | tw | ||
| (TpClPP)Cu 3a | 1.26 | 1.07 | 0.19 | −1.30 | −1.75 | 0.45 | 2.37 | 48 | ||
| Butano-(TpCH3PP)Cu 1b | 0.95 | 0.42 | 0.53 | −1.56 | −1.97 | 0.41 | 1.98 | tw | ||
| Butano-(TPP)Cu 2b | 0.97 | 0.49 | 0.48 | −1.55 | −1.89 | 0.34 | 2.04 | tw | ||
| Butano-(TpClPP)Cu 3b | 1.00 | 0.58 | 0.42 | −1.52 | −1.82 | 0.30 | 2.10 | tw | ||
| Benzo-(TpCH3PP)Cu 1c | 0.85 | 0.53 | 0.32 | −1.23 | −1.85 | 0.62 | 1.71 | tw | ||
| Benzo-(TPP)Cu 2c | 0.86 | 0.55 | 0.31 | −1.22 | −1.82 | 0.60 | 1.77 | tw | ||
| Benzo-(TpClPP)Cu 3c | 0.90 | 0.63 | 0.27 | −1.18 | −1.75 | 0.57 | 1.81 | tw | ||
| Br8(TPP)Cu | 1.41 | 0.96 | 0.45 | −0.87 | −1.12 | 0.25 | 1.83 | 48 | ||
| (DPP)Cu | 1.00 | 0.55 | 0.45 | −1.32 | −1.60 | 0.28 | 1.87 | 49 | ||
| (OETPP)Cu | 0.97 | 0.38 | 0.59 | 52 | ||||||
| PhCNb | Butano-(TpCH3PP)Cu 1b | 1.99a | 0.98 | 0.52 | 0.46 | −1.51 | −1.94a | 0.43 | 2.03 | tw |
| Butano-(TPP)Cu 2b | 2.00a | 0.98 | 0.54 | 0.44 | −1.48 | −1.92a | 0.44 | 2.02 | tw | |
| Benzo-(TPP)Cu 2c | 1.88a | 0.89 | 0.60 | 0.29 | −1.18 | −1.78a | 0.60 | 1.78 | tw | |
| (TPP)Cu | 1.33 | 1.03 | 0.30 | −1.26 | −1.72 | 0.46 | 2.29 | 12 | ||
| (OETPP)Cu | 2.00a | 0.97 | 0.46 | 0.51 | −1.46 | −1.90 | 0.44 | 1.92 | 12 | |
| (DPP)Cu | 1.88 | 0.94 | 0.54 | 0.40 | −1.22 | −1.61 | 0.39 | 1.76 | 12 | |
| DMFc | (TpCH3PP)Cu 1a | 1.18 | 0.99 | 0.19 | −1.12 | −1.77a | 0.65 | 2.11 | tw | |
| (TPP)Cu 2a | 1.19 | 1.02 | 0.17 | −1.15 | −1.70 | 0.55 | 2.17 | tw | ||
| (TpClPP)Cu 3a | 1.24 | 1.09 | 0.15 | −1.10 | −1.60 | 0.50 | 2.19 | tw | ||
| Butano-(TpCH3PP)Cu 1b | 0.92 | 0.56 | 0.36 | −1.40 | −1.87a | 0.47 | 1.96 | tw | ||
| Butano-(TPP)Cu 2b | 0.92 | 0.59 | 0.33 | −1.40 | −1.83a | 0.43 | 1.99 | tw | ||
| Butano-(TpClPP)Cu 3b | 0.99 | 0.66 | 0.33 | −1.31 | −1.74a | 0.43 | 1.97 | tw | ||
| Benzo-(TPP)Cu 2c | 0.82 | 0.61 | 0.21 | −1.10 | −1.66 | 0.56 | 1.71 | tw | ||
| Benzo-(TpClPP)Cu 3c | 0.86 | 0.67 | 0.19 | −1.03 | −1.66a | 0.63 | 1.70 | tw | ||
 | ||
| Fig. 4 Cyclic voltammograms showing oxidations of (a) butanoporphyrins 1b–3b and (b) benzoporphyrins 1c–3c in CH2Cl2 containing 0.1 M TBAP. | ||
As compared to the three butano-(TpYPP)CuII derivatives, the first oxidation of the corresponding benzo-(TpYPP)CuII derivatives with the same meso-substituents is harder by 50–100 mV while the second oxidation of the benzoporphyrins is easier by 100–110 mV. This is seen in Fig. 3 for compounds 2b and 2c in CH2Cl2 and also in Fig. 4b where ΔE1/2 between the first two oxidations of 1c–3c in CH2Cl2 ranges from 0.32 V for Y = CH3 to 0.27 V for Y = Cl. These separations are slightly larger than the ΔE1/2 of the corresponding (TpYPP)Cu compounds, as seen in Table 2.
For all of the examined tetrabenzoporphyrins, the separation, ΔE(2o−1o) of compounds 1c–3c is much smaller than that of the tetrabutanoporphyrins 1b–3b. However, it should be pointed out that the nature of the para-substituent on the meso-phenyl rings also affects the potential separations of compounds in all three series. The largest values of ΔE(2o−1o) are seen for the porphyrins having an electron-donating para-CH3 substituent on the four meso-phenyl groups while the smallest ΔE(2o−1o) are seen for the porphyrins with an electron-withdrawing Cl group on the phenyl rings of the macrocycle.
Thin-layer UV-visible spectroelectrochemistry was carried out during the two oxidations of each porphyrin in CH2Cl2 containing 0.1 M TBAP. Examples of the spectral changes are illustrated in Fig. 5 for compounds 2a, 2b and 2c in CH2Cl2. The applied oxidation potentials are indicated in the figure and were in each case sufficiently positive of E1/2 for the specific redox couple to guarantee a complete conversion of the neutral porphyrin to its singly or doubly oxidized form.
 | ||
| Fig. 5 Thin-layer UV/Vis spectral changes of (a) (TPP)CuII 2a, (b) butano-(TPP)CuII 2b and (c) benzo-(TPP)CuII 2c during the first and second controlled potential oxidations in CH2Cl2, 0.1 M TBAP. Cyclic voltammograms for the compounds are shown in Fig. 4. | ||
As seen in the figure, the three porphyrins exhibit similar spectral changes independent of the β,β′-fused groups. The Soret and Q bands both decrease in intensity during the first oxidation of each compound (see spectra at top of Fig. 5), indicating formation of porphyrin π-cation-radicals under the given solution conditions. The spectral changes during the second oxidation (bottom of Fig. 5) indicate the formation of a porphyrin dication.
It was long thought that Cu(II) porphyrins would undergo only porphyrin macrocycle-centered oxidations in nonaqueous media1,2 but a metal-centered oxidation to give a Cu(III) porphyrin was recently demonstrated as being possible for the nonplanar copper porphyrins (OETPP)Cu and (DPP)Cu in PhCN containing 0.1 M TBAP.12 In the case of the currently investigated copper tetrabutanoporphyrins and tetrabenzoporphyrins, a metal-centered oxidation is also observed under the same solution conditions. Examples are given in Fig. 6 for compounds 1b, 2b and 2c, where the CuII/CuIII process is located at ∼2.00 V vs. SCE in PhCN. The CuII/CuIII transitions of (DPP)Cu and (OETPP)Cu are located at similar potentials in this solvent (see Fig. 6a and b).
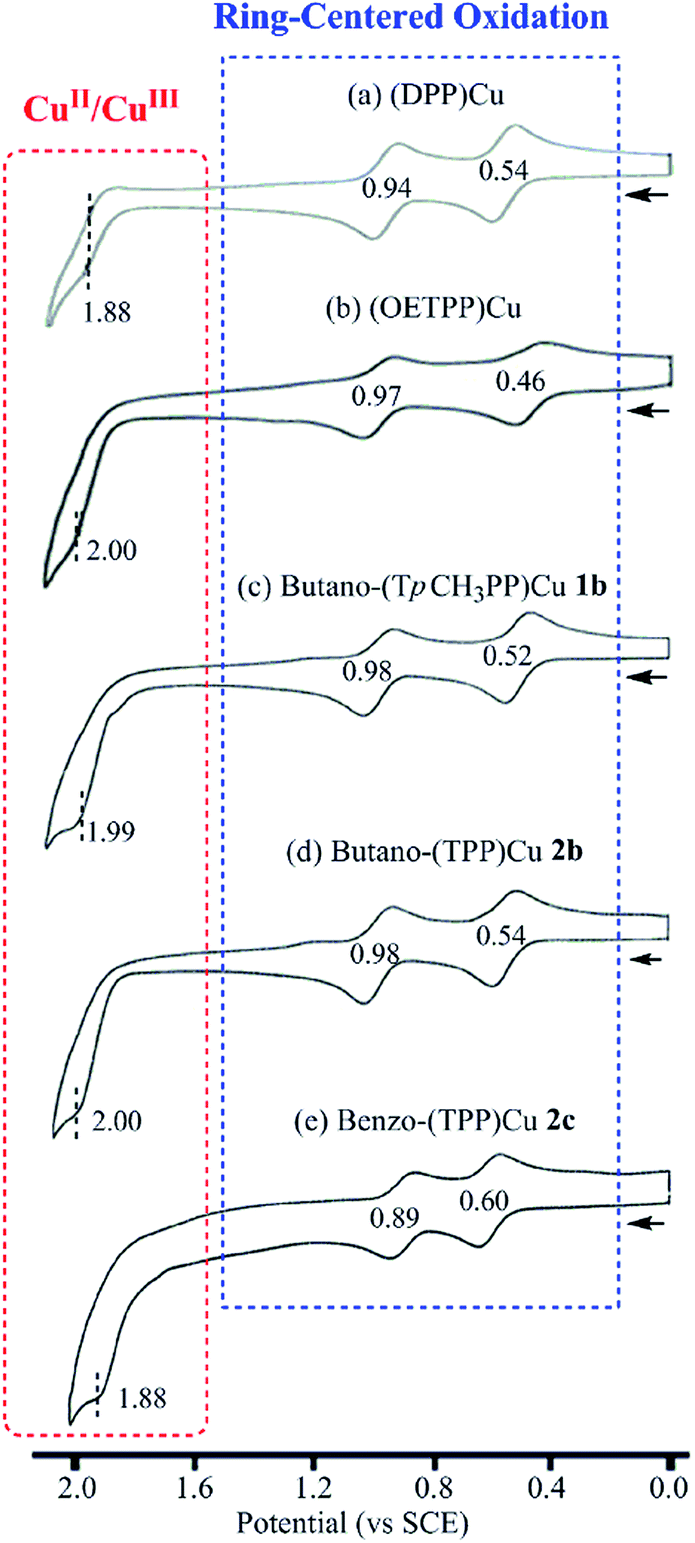 | ||
| Fig. 6 Cyclic voltammograms of (a) (DPP)Cu, (b) (OETPP)Cu, (c) butano-(TpCH3PP)Cu 1b, (d) butano-(TPP)Cu 2b and (e) benzo-(TPP)Cu 2c in PhCN containing 0.1 M TBAP. Scan rate = 0.1 V s−1. | ||
 | ||
| Fig. 7 Cyclic voltammograms showing reductions of the (a) tetrabutanoporphyrins 1b–3b and (b) tetrabenzoporphyrins 1c–3c in CH2Cl2 containing 0.1 M TBAP. | ||
Like the tetrabutanoporphyrins, the copper(II) tetrabenzoporphyrins 1c–3c also undergo two one-electron reductions, but the first reduction is shifted positively by over 300 mV as compared to that of the corresponding tetrabutanoporphyrins 1b–3b (see Fig. 3 and 7). This contrasts with the second reduction which occurs at very similar E1/2 values of −1.75 and −1.85 V. This difference between the two series of compounds leads to a much larger potential separation (0.57–0.62 V) between the first and second reductions of 1c–3c as compared to 1b–3b.
Fig. 8 illustrates the spectral changes for 2a, 2b and 2c during the first and second one-electron reductions in CH2Cl2, 0.1 M TBAP. As expected, the Soret and Q bands both decrease in intensity upon the stepwise one-electron additions, indicating formation of a π-anion radical and dianion in these processes. Similar spectral changes have been reported for other investigated Cu(II) porphyrins which are converted to their π-anion radical and dianion form in nonaqueous media.50
 | ||
| Fig. 8 Thin-layer UV/Vis spectral changes of (a) (TPP)CuII 2a, (b) butano-(TPP)CuII 2b and (c) benzo-(TPP)CuII 2c during the first and second controlled potential reductions in CH2Cl2, 0.1 M TBAP. Cyclic voltammograms of the compounds are shown in Fig. 7. | ||
Substituent effect
It has long been known that modification of a porphyrin structure by addition of electron-withdrawing or electron-donating substituents on the meso- and/or β-positions of the macrocycle may have a significant effect on the electrochemical properties.1,2 This is also the case for the currently investigated β,β′-fused butano- and benzoarylporphyrins.Fig. 9 shows how the meso-phenyl ring substituents effect redox potentials for reduction and oxidation of the butano-(TpYPP)Cu and benzo-(TpYPP)Cu compounds in CH2Cl2. A linear relationship is observed between the measured E1/2 values for electron addition or electron abstraction and the sum of the Hammett substituent constants defined by σ.51 The magnitude of the substituent effect is given by the slope of the correlation using the relationship ΔE1/2 = Σσρ, where ρ is the reaction constant which measures the magnitude of the substituent effect on the redox potentials.51
 | ||
| Fig. 9 Plot of reduction and oxidation potentials in CH2Cl2 vs. the sum of Hammett constants (Σσ) of (a) butano-(TpYPP)Cu 1b–3b and (b) benzo-(TpYPP)Cu 1c–3c. | ||
As seen in Fig. 9, the calculated ρ values for the first reduction of 1b–3b and 1c–3c are 25 and 32 mV, respectively, and both values are much smaller than the ρ of 93 and 83 mV (the slope of the correlation) for the second reduction of the same compounds. This result indicates that the meso-substituents have a larger effect on the second reduction than the first reduction of the compounds. In contract, the first oxidations of compounds in the two series have larger ρ values (99 mV for 1b–3b and 64 mV for 1c–3c) than the second oxidations where the slopes (ρ values) are 31 and 32 mV, respectively.
The magnitude of the substituent effect for oxidation of the Cu(II) tetrabenzo- and tetrabutanoporphrins is virtually identical to earlier measured substituent effects for oxidation of Cu(II) tetrarylporphyrins with the same type of meso-phenyl substituents.53 This is significant in that it indicates a similar site of oxidation, independent of the porphyrin ring planarity, the degree of β-pyrrole substitution or the addition of four fused π-ring systems to the macrocycle.
The β,β′-fused tetrabutano and tetrabenzo groups have a significant effect not only on the individual reduction and oxidation potentials but also on the HOMO–LUMO gap of the porphyrins. This is shown by the cyclic voltammograms in Fig. 3 for 2a, 2b and 2c in CH2Cl2, 0.1 M TBAP. The first reduction of benzo-(TPP)CuII 2c (E1/2 = −1.22 V) is easier than the first reduction of 2a by only 60 mV but the first two oxidations exhibit very large negative shifts (490–440 mV) as compared to the E1/2 values for the same two redox reactions of (TPP)CuII 2a in CH2Cl2. This is due to the effect of the extended π-system and the known distortion of the macrocycle of 2c.32 The first oxidation of butano-(TPP)CuII 2b is easier than the first oxidation of benzo-(TPP)CuII 2c, indicating that a stronger distortion might occur for compound 2b than for 2c. The first reduction of 2b is much more difficult to reduce than 2a, which is consistent with the electron-donating β,β′-fused butano-substituents on 2b which lead to a harder reduction at the conjugated π-ring system of the macrocycle.
Finally, it should be pointed out that among the three series of investigated compounds, the tetrabenzotetraaryl-porphyrins have the smallest HOMO–LUMO gap which averages 1.76 ± 0.05 V in CH2Cl2, 1.78 V (2c) in PhCN and 1.70 ± 0.01 V (2c, 3c) in DMF. Similar HOMO–LUMO gaps are also seen for the two nonplanar copper porphyrins, (OETPP)Cu and (DPP)Cu, both of which exhibit a CuII/CuIII process in PhCN.12 The HOMO–LUMO gap of the investigated tetrabutanoporphyrins averages 2.04 ± 0.06 V while the average gap of the T(pYPP)Cu derivatives is 2.33 ± 0.03 V in CH2Cl2.
Experimental
Chemicals
Dichloromethane (CH2Cl2, 99.8%) was purchased from EMD Chemicals Inc. and used as received. N,N-Dimethylformamide (DMF) was purchased from Sigma-Aldrich and used as received. Tetra-n-butylammonium perchlorate (TBAP) was purchased from Sigma Chemical or Fluka Chemika Co. and used as received.Instrumentation
Cyclic voltammetry was carried out at 298 K using an EG&G Princeton Applied Research (PAR) 173 potentiostat/galvanostat. A homemade three-electrode cell was used for cyclic voltammetric measurements. It consisted of a glassy carbon working electrode, a platinum counter electrode, and a homemade saturated calomel reference electrode (SCE). The SCE was separated from the bulk of the solution by a fritted glass bridge of low porosity, which contained the solvent/supporting electrolyte mixture. UV/Vis spectra were measured using a Hewlett–Packard 8453 diode array spectrophotometer.Thin-layer UV/Vis spectroelectrochemical experiments were performed with a home-built thin-layer cell which has a light transparent platinum net working electrode. Potentials were applied and monitored with an EG&G PAR model 173 potentiostat. Time-resolved UV/Vis spectra were recorded with a Hewlett–Packard model 8453 diode array spectrophotometer. High purity N2 was used to deoxygenate the solution and kept over the solution during each electrochemical and spectroelectrochemical experiment. MALDI-TOF mass spectra were carried out on a Bruker BIFLEX III ultrahigh resolution. ESR spectra were recorded on a Bruker 300C spectrometer.
Synthesis of (TpYPP)Cu 1a–3a
The (TpYPP)4Cu derivatives were synthesized and purified according to a published literature procedure.31(TpCH3PP)Cu 1a (30 mg, 87% yield). UV/Vis (CH2Cl2): λmax, 418, 540 nm. MS (MALDI-TOF): m/z for C48H36CuN4, calcd, 731.224, found, 730.975.
(TPP)Cu 2a (38 mg, 90% yield). UV/Vis (CH2Cl2): λmax, 416, 540 nm. MS (MALDI-TOF): m/z for C44H28CuN4, calcd, 675.161, found, 674.909.
(TpClPP)Cu 3a (35 mg, 85% yield). UV/Vis (CH2Cl2): λmax, 415, 539 nm. MS (MALDI-TOF): m/z for C44H24Cl4CuN4, calcd, 811.005, found, 811.004.
Synthesis of copper tetrabutanotetraarylporphyrins 1b–3b
Free-base butano-(TpYPP)H2 (∼100 mg), which was synthesized according to a procedure in the literature,30,31 was dissolved in 50 mL CHCl3/MeOH (v/v 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) containing excess Cu(OAc)2·H2O (300 mg). The mixture was stirred at room temperature for half a day and the progress of the reaction was monitored by TLC and UV-Vis spectroscopy. After the starting compound was completely consumed, the mixture was evaporated to dryness and then freshly chromatographed with neutral alumina (200–300 mesh) using CH2Cl2 as eluent. The brown-red fraction was collected and evaporated to dryness.
:1) containing excess Cu(OAc)2·H2O (300 mg). The mixture was stirred at room temperature for half a day and the progress of the reaction was monitored by TLC and UV-Vis spectroscopy. After the starting compound was completely consumed, the mixture was evaporated to dryness and then freshly chromatographed with neutral alumina (200–300 mesh) using CH2Cl2 as eluent. The brown-red fraction was collected and evaporated to dryness.
Butano-(TpCH3PP)Cu 1b (86 mg, 80% yield). UV/Vis (CH2Cl2): λmax, 428, 560 nm. MS (MALDI-TOF): m/z for C64H60CuN4, calcd, 947.411, found, 947.410.
Butano-(TPP)Cu 2b (91 mg, 85% yield). UV/Vis (CH2Cl2): λmax, 426, 558 nm. MS (MALDI-TOF): m/z for C60H52CuN4, calcd, 891.349, found, 891.347.
Butano-(TpClPP)Cu 3b (88 mg, 83% yield). UV/Vis (CH2Cl2): λmax, 426, 559 nm. MS (MALDI-TOF): m/z for C60H48Cl4CuN4, calcd, 1027.193, found, 1030.239.
Synthesis of copper tetrabenzotetraarylporphyrins 1c–3c31
The butano-(TpYPP)Cu derivatives (∼60 mg) were dissolved in 150 mL distilled dry THF and DDQ was added to the solution which was kept under reflux for 1 h as the color of the solution changed from red to deep green. The reaction mixture was allowed to cool, diluted with CH2Cl2 and washed with water and NaCl solution. The solvent was removed under vacuum and the residue was purified on a silica gel column with CH2Cl2 as eluent. The first dark-green fraction was collected and evaporated to dryness. The resulting blue-green powder was the pure product.Benzo-(TpCH3PP)Cu 1c (28 mg, 48% yield). UV/Vis (CH2Cl2): λmax, 448, 462, 597, 648 nm. MS (MALDI-TOF): m/z for C64H44CuN4, calcd, 931.286, found, 933.289.
Benzo-(TPP)Cu 2c (32 mg, 55% yield). UV/Vis (CH2Cl2): λmax, 448, 460, 597, 648 nm. MS (MALDI-TOF): m/z for C60H36CuN4, calcd, 875.224, found, 875.222.
Benzo-(TpClPP)Cu 3c (26 mg, 45% yield). UV/Vis (CH2Cl2): λmax, 447, 459, 600, 649 nm. MS (MALDI-TOF): m/z for C60H32Cl4CuN4, calcd, 1011.068, found, 1016.026.
Conclusions
The four β,β′-fused butano or benzo-rings on the copper porphyrins have a significant effect on UV-Vis spectra, reduction–oxidation potentials and the HOMO–LUMO gap of the investigated compounds. A 10–20 nm red-shifted Soret band is seen for the tetrabutanoporphyrins and a 40–110 nm red-shift in the Soret band of the tetrabenzoporphyrins as compared to the related non-β-substituted compounds. A Cu(II)/Cu(III) conversion was observed for the tetrabutano- and tetrabenzo-porphyrins in PhCN, and this is attributed to the nonplanarity of porphyrin macrocycle. As is also the case for the fully β-substituted nonplanar copper porphyrins, (OETPP)Cu and (DPP)Cu, the related β,β′-fused butano- and β,β′-fused benzoporphyrins with Cu(II) metal ions have a HOMO–LUMO gap of 2.04 ± 0.06 V and 1.76 ± 0.05 V, respectively in nonaqueous media.Acknowledgements
We gratefully acknowledge support from the Natural Science Foundation of China (Grant No. 21071067), Jiangsu University Foundation (Grant No. 05JDG051) and the Robert A. Welch Foundation (K. M. K., E-680).Notes and references
- K. M. Kadish, E. van Caemelbecke and G. Royal, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, San Diego, 2000, vol. 8, pp. 1−114 Search PubMed.
- K. M. Kadish, Prog. Inorg. Chem., 1986, 34, 435–605 CrossRef CAS PubMed.
- B. Sun, Z. P. Ou, S. B. Yang, D. Y. Meng, G. F. Lu, Y. Y. Fang and K. M. Kadish, Dalton, Trans., 2014, 43, 10809–10815 RSC.
- B. Sun, Z. P. Ou, D. Y. Meng, Y. Y. Fang, Y. Song, W. H. Zhu, P. V. Solntsev, V. N. Nemykin and K. M. Kadish, Inorg. Chem., 2014, 53, 8600–8609 CrossRef CAS PubMed.
- S. B. Yang, B. Sun, Z. P. Ou, D. Y. Meng, G. F. Lu, Y. Y. Fang and K. M. Kadish, J. Porphyrins Phthalocyanines, 2013, 17, 857–869 CrossRef CAS.
- A. X. Lv, Y. Y. Fang, M. Zhu, S. Huang, Z. P. Ou and K. M. Kadish, J. Porphyrins Phthalocyanines, 2012, 16, 310–315 CrossRef.
- S. Fukuzumi, K. Ohkubo, W. H. Zhu, M. Sintic, T. Khoury, P. J. Sintic, W. E, Z. P. Ou, M. J. Crossley and K. M. Kadish, J. Am. Chem. Soc., 2008, 130, 9451–9458 CrossRef CAS PubMed.
- T. D. Lash, D. A. Colby and L. F. Szczepura, Inorg. Chem., 2004, 43, 5258–5267 CrossRef CAS PubMed.
- I. Zilbermann, J. Hayon, E. Maimon, R. Ydgar, E. Korin and A. Bettelheim, Electrochem. Commun., 2002, 4, 862–865 CrossRef CAS.
- Z. P. Ou, T. Khoury, Y. Y. Fang, W. H. Zhu, P. J. Sintic, M. J. Crossley and K. M. Kadish, Inorg. Chem., 2013, 52, 2474–2483 CrossRef CAS PubMed.
- Z. P. Ou, W. H. Zhu, Y. Y. Fang, P. J. Sintic, T. Khoury, M. J. Crossley and K. M. Kadish, Inorg. Chem., 2011, 50, 12802–12809 CrossRef CAS PubMed.
- Y. Y. Fang, M. O. Senge, E. van Caemelbecke, K. M. Smith, C. J. Medforth, M. Zhang and K. M. Kadish, Inorg. Chem., 2014, 53, 10772–10778 CrossRef CAS PubMed.
- A. A. Sinelshchikova, S. E. Nefedov, Y. Y. Enakieva, Y. G. Gorbunova, A. Y. Tsivadze, K. M. Kadish, P. Chen, A. Bessmertnykh-Lemeune, C. Stern and R. Guilard, Inorg. Chem., 2013, 52, 999–1008 CrossRef CAS PubMed.
- S. Mirra, S. Milione, M. Strianese and C. Pellecchia, Eur. J. Inorg. Chem., 2015, 2272–2276 CrossRef CAS PubMed.
- M. M. Yu, C. Wang, J. Li, L. Yuan and W. J. Sun, Appl. Surf. Sci., 2015, 342, 47–54 CrossRef.
- T. Nguyen, P. Hakansson, R. Edge, D. Collison, B. A. Goodman, J. R. Burns and E. Stulz, New J. Chem., 2014, 38, 5254–5259 RSC.
- M. M. Yu, J. Li, W. J. Sun, M. Jiang and F. X. Zhang, J. Mater. Sci., 2014, 49, 5519–5528 CrossRef.
- T. I. S. Oliveira, V. N. dos Santos, D. Lomonaco, A. N. Correia, S. E. Mazetto and P. de Lima-Neto, J. Electrochem. Soc., 2013, 160, B113–B118 CrossRef.
- Y. Y. Liu, Y. M. Yang, Q. L. Sun, Z. Y. Wang, B. B. Huang, Y. Dai, X. Y. Qin and X. Y. Zhang, ACS Appl. Mater. Interfaces, 2013, 5, 7654–7658 CAS.
- P. Hakansson, T. Nguyen, P. B. Nair, R. Edge and E. Stulz, Phys. Chem. Chem. Phys., 2013, 15, 10930–10941 RSC.
- M. H. Ha-Thi, N. Shafizadeh, L. Poisson and B. Soep, J. Phys. Chem. A, 2013, 117, 8111–8118 CrossRef CAS PubMed.
- G. P. Yao, J. Li, Y. Luo and W. J. Sun, J. Mol. Catal. A: Chem., 2012, 361–362, 29–35 CrossRef CAS PubMed.
- G. Q. Liu, A. N. Khlobystov, G. Charalambidis, A. G. Coutsolelos, G. A. D. Briggs and K. Porfyrakis, J. Am. Chem. Soc., 2012, 134, 1938–1941 CrossRef CAS PubMed.
- W. J. Sun, J. Li, G. P. Yao, M. Jiang and F. X. Zhang, Catal. Commun., 2011, 16, 90–93 CrossRef CAS PubMed.
- G. V. Ponomarev, D. V. Yashunsky and D. P. Arnold, Recent Res. Dev. Pure Appl. Chem., 1998, 2(1), 199–215 CAS.
- M. S. Asano, K. Okamura, A. Jin-mon, S. Takahashi and Y. Kaizu, Chem. Phys., 2013, 419, 250–260 CrossRef CAS PubMed.
- S. Afzal, W. A. Daoud and S. J. Langford, ACS Appl. Mater. Interfaces, 2013, 5, 4753–4759 CAS.
- X. Q. Su, J. Li, Z. Q. Zhang, M. M. Yu and L. Yuan, J. Alloys Compd., 2015, 626, 252–259 CrossRef CAS PubMed.
- W. J. Sun, J. Li, G. Mele, Z. Q. Zhang and F. X. Zhang, J. Mol. Catal. A: Chem., 2013, 366, 84–91 CrossRef CAS PubMed.
- C. M. B. Carvalho, T. J. Brocksom and K. Thiago de Oliveira, Chem. Soc. Rev., 2013, 42, 3302–3317 RSC.
- O. S. Finikova, A. V. Cheprakov, I. P. Beletskaya, P. J. Carroll and S. A. Vinogradov, J. Org. Chem., 2004, 69, 522–535 CrossRef CAS.
- L. D. Sparks, C. J. Medforth, M. S. Park, J. R. Chamberlain, M. R. Ondrias, M. O. Senge, K. M. Smith and J. A. Shelnutt, J. Am. Chem. Soc., 1993, 115, 581–592 CrossRef CAS.
- D. Chumakov, A. Moiseeva, A. Anisimov, B. Uzhinov and A. Khoroshutin, J. Porphyrins Phthalocyanines, 2010, 14, 815–824 CrossRef CAS.
- M. H. Qi and G. F. Liu, J. Porphyrins Phthalocyanines, 2004, 8, 1187–1195 CrossRef CAS.
- K. M. Kadish, O. S. Finikova, E. Espinosa, C. P. Gros, G. de Stefano, A. V. Cheprakov, I. P. Beletskaya and R. Guilard, J. Porphyrins Phthalocyanines, 2004, 8, 1062–1066 CrossRef CAS.
- P. Chen, O. S. Finikova, Z. P. Ou, S. A. Vinogradov and K. M. Kadish, Inorg. Chem., 2012, 51, 6200–6210 CrossRef CAS PubMed.
- V. V. Rozhkov, M. Khajehpour and S. A. Vinogradov, Inorg. Chem., 2003, 42, 4253–4255 CrossRef CAS PubMed.
- J. E. Rogers, K. A. Nguyen, D. C. Hufnagle, D. G. McLean, W. J. Su, K. M. Gossett, A. R. Burke, S. A. Vinogradov, R. Pachter and P. A. Fleitz, J. Phys. Chem. A, 2003, 107, 11331–11339 CrossRef CAS.
- G. Dougherty and R. F. Pasternack, Inorg. Chim. Acta, 1992, 195, 95–100 CrossRef CAS.
- A. Pezeshk, J. Phys. Chem., 1990, 94, 451–456 CrossRef CAS.
- C. Mengersen, J. Subramanian and J.-H. Fuhrhop, Mol. Physiol., 1976, 32, 893–897 CrossRef CAS PubMed.
- M. Ravikanth, D. Reddy, A. Misra and T. K. Chandrashekar, J. Chem. Soc., Dalton Trans., 1993, 1137–1141 RSC.
- L. Banci, Inorg. Chem., 1985, 24, 782–786 CrossRef CAS.
- S. P. Greiner, D. L. Rowlands and R. W. Kreilick, J. Phys. Chem., 1992, 96, 9132–9139 CrossRef CAS.
- S. Konishi, M. Hoshino and M. Imamura, Chem. Phys. Lett., 1983, 94, 267–269 CrossRef CAS.
- J. A. de Bolfo, T. D. Smith, J. F. Boas and J. R. Pilbrow, J. Chem. Soc., Dalton Trans., 1975, 1523–1525 RSC.
- P. D. W. Boyd, T. D. Smith, J. H. Price and J. R. Pilbrow, J. Phys. Chem., 1972, 56, 1253–1263 CrossRef CAS PubMed.
- G. Hariprasad, S. Dahal and B. G. Maiya, J. Chem. Soc., Dalton Trans., 1996, 3429–3436 RSC.
- J. Takeda and M. Sato, Chem. Lett., 1995, 939–940 CrossRef CAS.
- K. M. Kadish, C. Araullo, G. B. Maiya, D. Sazou, J.-M. Barbe and R. Guilard, Inorg. Chem., 1989, 28, 2528–2533 CrossRef CAS.
- P. Zuman, in Substituent Effects in Organic Polarography, Plenum Press, New York, 1967 Search PubMed.
- M. W. Renner, K. M. Barkigia, Y. Zhang, C. J. Medforth, K. M. Smith and J. Fajer, J. Am. Chem. Soc., 1994, 116, 8582–8592 CrossRef CAS.
- K. M. Kadish and M. M. Morrison, Bioinorg. Chem., 1977, 7, 107–115 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |