
A pH-sensitive nanocarrier for co-delivery of doxorubicin and camptothecin to enhance chemotherapeutic efficacy and overcome multidrug resistance in vitro
Abstract
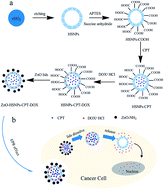
To deliver chemotherapeutic drugs simultaneously, using drug carriers is a feasible strategy for a better synergetic effect. We have constructed hollow silica nanoparticles (HSNPs) sealed with ZnO quantum dots (QDs) as a pH-sensitive nanocarrier, where the HSNPs have large hollow interiors for delivering hydrophobic camptothecin (CPT) and a mesoporous structure for delivering hydrophilic doxorubicin (DOX·HCl). This cooperative drug loading has largely increased the drug encapsulation efficiency of both CPT and DOX up to 89.28% and 44.98%, respectively. Meanwhile, the ZnO QD lids in this drug delivery system are found to be rapidly dissolved in the acidic intracellular compartments to trigger pH-sensitive drug release. The co-delivery of multi-drugs with different anticancer mechanisms in the same nanocarrier enables the intracellular release of the drug combinations to greatly enhance chemotherapeutic efficacy and overcome multidrug resistance (MDR).
 Please wait while we load your content...
Please wait while we load your content...

