
Bio-compatible poly(ester-urethane)s based on PEG–PCL–PLLA copolymer with tunable crystallization and bio-degradation properties
Guangzhong Yinad,
Donglin Zhaoabd,
Xiao Wangc,
Ye Renad,
Lianwei Zhangad,
Xingxin Wuc,
Shaoping Niec and
Qifang Li*abd
aState Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: qflee@mail.buct.edu.cn
bKey Laboratory of Carbon Fiber and Functional Polymers, Ministry of Education, Beijing University of Chemical Technology, Beijing 100029, China
cEmergency & Critical Care Center, Beijing Anzhen Hospital, Capital Medical University, Beijing, 100029, China
dCollege of Material Science and Engineering, Beijing University of Chemical Technology, Beijing 100029, China
First published on 8th September 2015
Abstract
Multi-block copolymer poly(ester-urethane)s with ethoxy or polyhedral oligomeric silsesquioxane (POSS) terminal functional groups were designed and synthesized by using polyethylene glycol–poly(ε-caprolactone)–poly(L-lactide) (PEG–PCL–PLLA) diols via urethane linkages. In addition, an efficient and green catalyst, bismuth ethylhexanoate, was used to prepare high molecular weight copolymers, and the influences of molecular compositions on the crystallinity, mechanical properties, biodegradability, cytocompatibility and blood-compatibility were systematically studied. By varying the terminal functional groups, as well as PEG, PCL and PLLA segment ratios, the materials exhibited highly tunable properties, especially including crystallinity, mechanical properties and degradation rate. Notably, these new functional materials may effectively be applied in the treatment of blood vessels because of the mentioned tunable properties.
Introduction
Poly(ε-caprolactone) (PCL) and poly(L-lactide) (PLLA) are of practical significance and great research value,1,2 and have been used widely as biodegradable polymer scaffolds for implantation of human tissues, e.g. for vascularized tracheal substitute,3 bone formation,4,5 cartilage regeneration matrix,6 and so on. An ideal tissue engineering material should be able to mimic the structure of the native extracellular matrix, so as to provide mechanical support and regulate cellular activities. Generally, pure PCL or PLLA can't meet the functional requirements well. To address these limitations, several works have been performed to modify the functionality of polymer matrix. Some chemical methods were reported recently, for examples, Melchiorri et al. reported surface modification of PCL scaffold using Heparin,7 and Huang et al. prepared P(LLA–CL) copolymer to be an artificial blood vessel.8 Numerous studies were mainly focused on technical level, modification via blending by electrospinning technique,9–13 surface modification of electrospun membranes14,15 or other technics.16,17 For examples, Kim et al.16 prepared PCL/alginate composite scaffolds multilayered 3D structure, which have potential for use in scaffolds for hard tissue regeneration. Zhang et al.17 successfully accomplished PCL scaffolds by solvent casting/salt leaching and thermal induced phase separation (TIPS) methods, and genipin-cross-linked gelatin entrapped with recombinant human BMP-2 was exploited to decorate the interior surface of the scaffolds.Biomaterials with tunable mechanics and controllable degradation rates should be taken into an important consideration when designing tissue engineering materials, because the degradation rate should ideally match the regeneration rate of cells and tissues.18,19 As well known, PCL presented slow degradation rate20 and excellent flexibility; the PLLA was brittle materials, but with a faster degradation rate.21 In principle, control over mechanical properties and degradation rate can be sought through the preparation of novel copolymers covering a range of compositions. Indeed, the PCL–PLLA-based copolymers or the corresponding poly(ester-urethane)s were recently reported,22 especially, Peponi et al.23,24 synthesized tremendous related polymers varying both the molecular weight of the PCL and PLLA blocks as well as the relative content of each block in the copolymer, focusing on the relationship between their chemical compositions and their tailored final properties.
Polyethylene glycol (PEG) was widely used to improve the bio-compatibility of the blood contacting materials.25 In addition, polyhedral oligomeric silsesquioxanes (POSS), a class of hybrid molecules with an inorganic silicon oxygen cage,26–28 which can generally improve thermal stability and mechanical properties, had good biocompatibility.29 Mather et al.30–32 reported the existence of POSS can effectively regulate the degradation of the material properties, because it can effectively regulate water absorption.33 To achieve the performance requirements, PEG, PCL, PLLA and POSS were incorporated together in molecular level, resulting in liner PEG–PCL–PLLA-based tissue engineering materials (Scheme 1). It was expected that the materials would exhibit highly tunable properties, including crystallinity, mechanical properties, controllable degradation rate and bio-compatibility. Notably, the current work is expected to provide valuable information for the development of new functional materials, which may effectively be applied in the treatment of blood vessels.
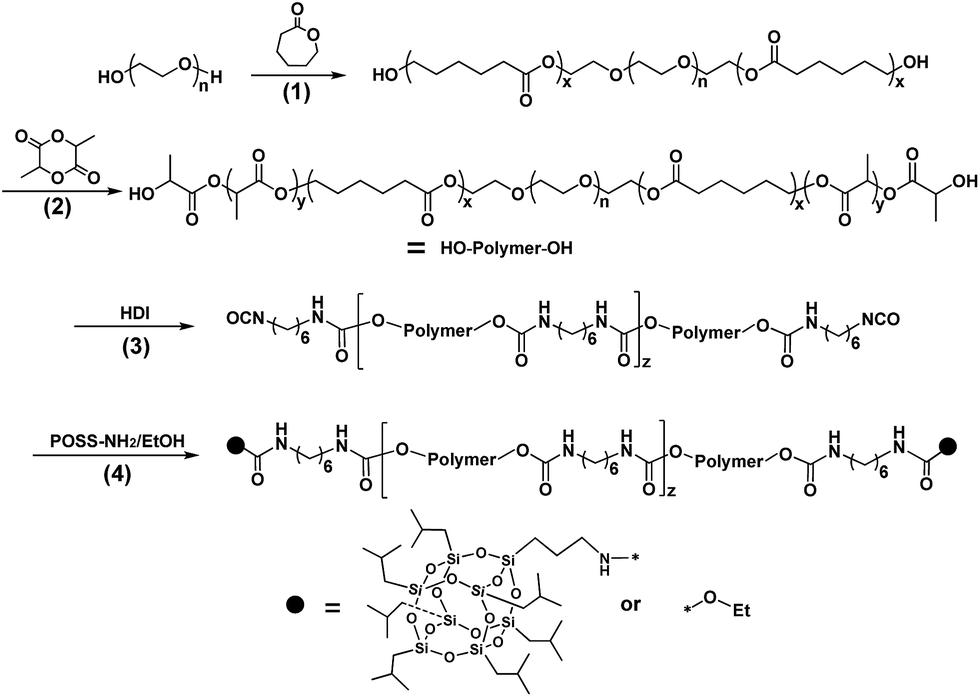 | ||
| Scheme 1 Synthesis of copolymers: step (1), the synthesis of PEG–PCL by ROP; step (2), synthesis of PEG–PCL–PLLA by ROP; step (3), chain extension by HDI; step (4), functional terminal. | ||
Experimental
Materials
ε-Caprolactone (ε-CL, Alfa Aesar) was purified by vacuum distillation over calcium hydride (CaH2). PEG 2000 (Alfa Aesar) was dried in vacuum oven for 48 h at 95 °C. Dichloromethane (DCM) and toluene were purified by distillation over CaH2 (Alfa Aesar). Stannous, 96% (Alfa Aesar), 1,8-diazabicyclo-[5.4.0]-undec-7-ene (DBU), 99% (J&K), L-(−)-lactide, 99% (J&K), hexa methylenediisocyanate (HDI), 99% (J&K), bismuth ethylhexanoate, 99% (J&K) and aminopropyllsobutyl POSS, 99%, (hybrid plastic) were used as received. Other solvents were obtained from Beijing Chemical Works and used without any treatment.The synthesis of PEG–PCL
The PEG–PCL polymer was synthesized by the ring-opening polymerization (ROP) of ε-caprolactone (ε-CL) using PEG as the initiator and stannous as the catalyst according to previous reports.34,35 Typically, PEG 2000 and ε-CL were weighed into eggplant-type reaction flask. Then 0.2–0.5 wt% of stannous was dropped into the mixture. The flask was immersed in an oil bath at 110 °C with magnetic stirring for 24 h under nitrogen purge. The product was dissolved in 20 mL CHCl3, and precipitated into n-hexane (400 mL). After filtration and drying at vacuum oven at 40 °C for 24 h, a white product was obtained (>95% yield, Mn ∼ 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 or 9800 g mol−1 by GPC). FTIR (cm−1, KBr window): 3439 (–OH), 2946–2867 (–CH2–CH2–), 1726 (C
000 g mol−1 or 9800 g mol−1 by GPC). FTIR (cm−1, KBr window): 3439 (–OH), 2946–2867 (–CH2–CH2–), 1726 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1189 (C–O–C). 1H NMR in CDCl3 (ppm): δ 4.245 (t, J = 4.85 Hz, –OCH2C
O), 1189 (C–O–C). 1H NMR in CDCl3 (ppm): δ 4.245 (t, J = 4.85 Hz, –OCH2C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2O(CO)–), δ 4.081 (t, J = 6.70 Hz, –CH2C2O(CO)–), δ 3.665 (s, –OC2CH2–), δ 2.327 (t, J = 7.52 Hz, –O(CO)–C2CH2–), δ 1.711–1.628 (m, –O(CO)–CH2C2–), 1.441–1.365 (m, –CH2C2CH2–).
2O(CO)–), δ 4.081 (t, J = 6.70 Hz, –CH2C2O(CO)–), δ 3.665 (s, –OC2CH2–), δ 2.327 (t, J = 7.52 Hz, –O(CO)–C2CH2–), δ 1.711–1.628 (m, –O(CO)–CH2C2–), 1.441–1.365 (m, –CH2C2CH2–).
The synthesis of PEG–PCL–PLLA
The PEG–PCL–PLLA was prepared according to previous reports.36 Briefly, (i) PEG–PCL was dissolved in CH2Cl2 together with a predetermined amount of L-(−)-lactide in an oven-dried round-bottomed flask containing a magnetic stir bar. (ii) DBU was added at a concentration of 10 μL mL−1. The solution was vigorously stirred under nitrogen purge. After 1.5 h, solid benzoic acid was added. As above, the PEG–PCL–PLLA was purified by precipitation twice into n-hexane from CH2Cl2 and dried at 50 °C under vacuum overnight. 1H NMR in CDCl3 (ppm): δ 5.252 (q, J = 7.05 Hz, –O–(CH3)C–CO–), δ 4.247 (t, J = 4.86 Hz, –OCH2C2O(CO)–), δ 4.083 (t, J = 6.68 Hz, –CH2C2O(CO)–), δ 3.668 (s, –OC2CH2–), δ 2.329 (t, J = 7.51 Hz, –O(CO)–C2CH2–), δ 1.712–1.629 (m, –O(CO)–CH2C2–, –CO–C(C3)H–O–), δ 1.442–1.364 (m, –CH2C2CH2–).
Chain extention
In a 100 mL of three necked flask, PEG–PCL–PLLA diols (10 g) were dissolved in toluene (50 mL), which had been dried with the aid of CaH2. Under the protection of a nitrogen purge, the flask was heated to 95 °C, and a 0.5 mL of HDI was added to the 20 wt% toluene solution. Several drops of bismuth ethylhexanoate catalyst were added through a syringe. The reaction was kept at 95 °C for 12 h under the nitrogen purge, and a distinct viscosity rise was observed. After the full reaction, we added an appropriate amount of HDI to make sure we obtained NCO-terminated products. The polymer solution was then precipitated into an excess of anhydrous n-hexane and ether (v/v = 1:1). FTIR (cm−1, KBr window): 2946.07–2867.24 (–CH2–CH2–), 2230 (–NCO), 1726 (CO), 1628–1530 (–CONH–), 1189 (C–O–C).
Functional terminal
The products mentioned above as well as a certain amount of POSS or ethanol were dissolved in three-necked flask by anhydrous chloroform, stirred for 5 h at 80 °C, after precipitating in 10 volumes of n-hexane twice, the products were dried in vacuum at 45 °C for 24 h. The samples were washed two times with hexane to remove unreacted POSS. FTIR (cm−1, KBr window): 2946–2867 (–CH2–CH2–), 1726 (CO), 1630–1534 (–CONH–), 1190 (C–O–C). 1H NMR in CDCl3 (ppm): δ 5.252 (q, J = 7.05 Hz, –O–(CH3)C–CO–), δ 4.247 (t, J = 4.86 Hz, –OCH2C2O(CO)–), δ 4.083 (t, J = 6.68 Hz, –CH2C2O(CO)–), δ 3.668 (s, –OC2CH2–), δ 3.18 (–C2NH–), δ 2.329 (t, J = 7.51 Hz, –O(CO)–C2CH2–), δ 1.712–1.629 (m, –O(CO)–CH2C2–, –CO–C(C3)H–O–), δ 1.442–1.364 (m, –CH2C2CH2–), δ 0.97 (d, J = 6.588 Hz, –C3 (POSS)), δ 0.62 (d, J = 7.018 Hz, –C– (POSS)).
Sample preparation
The obtained materials from synthesis and precipitation were cast from CHCl3 solution, as indicated in the following example: the polymer (5 g) was dissolved in CHCl3 (40 mL) and poured in a PTFE casting dish with the dimension of 10 cm × 10 cm, and then transferred to a chamber to allow for a slow solvent evaporation. Following 48 h, the films were put in a vacuum at 50 °C to remove any residual solvent. The resulting films were flexible and nearly 0.40 mm in thickness.Characterization
1H NMR spectra were recorded in CDCl3 at 400 MHz in the AV400 (BRUKER) at 25 °C with deuterium solvents and TMS as an internal reference. Fourier transform infrared spectroscopy (FTIR) was recorded with Bruker Tensor-27 Fourier transform infrared spectrometer using the KBr disk method. Gel permeation chromatography (GPC) was performed on the system (Water 1515, Isocratic HPLC Pump and Water 2414 Refractive Index Detector) to obtain the molecular weights and molecular weights distribution of the polymers at room temperature. Tetrahydrofuran was used as mobile phase with a flow rate of 1.0 mL min−1 with column temperature at 35 °C, and monodisperse polystyrene standard samples were used for calibration.Differential scanning calorimetry (DSC) was performed using a DSC 200 PC (NETZSCH) differential scanning calorimeter calibrated with indium. Glass transition temperature (Tg) and melting temperature (Tm) were measured according to the running conditions: the sample was heated from room temperature to 180 °C (Process I) or 100 °C (Process II) (first heating, 20 °C min−1), kept isothermally for 5 min, cooled down to −100 °C, and heated again to 180 °C (second heating, 10 °C min−1). Furthermore, the degree of crystallization for each block was calculated based on DSC according to the following equation:
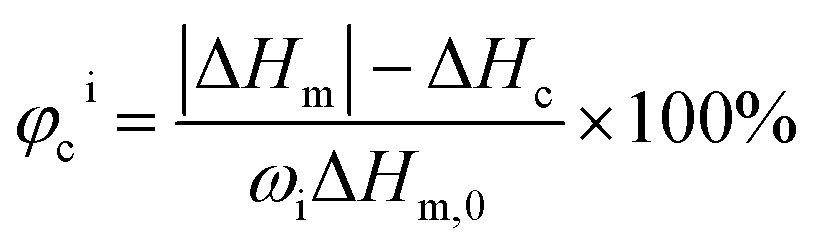 | (1) |
For optical microscopy observation, a Motic BA300 optical microscope equipped with a hot stage was used in this study.
The X-ray diffraction (XRD) data of the polymers were recorded with BRUKER D8 ADVANCE diffract meter using Cu Kα radiation from 5° to 40° at room temperature.
Uniaxial tensile tests were carried out at 25 °C with a crosshead speed of 50 mm min−1. Samples measuring 20 mm × 4 mm × 0.4 mm were cut from the films obtained above using a fresh razor blade. The modulus of each sample was determined by linearly fitting the elastic portion of the stress–strain curve before the yielding point. Five dog bone-shaped samples were analyzed.
In vitro degradation of all samples was carried out as reported elsewhere.37 Typically, preweighed samples were placed individually in test tubes containing phosphate buffered saline (PBS) at pH ∼ 7.4. The films were removed from the buffer solution and cleaned for three 15 min cycles each with deionized water under sonication. Thereafter, the cleaned films were freeze dried for 24 h. Mass remaining (MR%) was calculated according to the following equation:38
 | (2) |
MTT assay was used to test the cytotoxicity of the membranes. The 20 mm × 30 mm samples were sterilized by washing with a 75% (v/v) ethanol solution in sterilized water, exposed to Co60 for 15 min, and then incubated in DMEM at a proportion of 3 cm2 mL−1 for 24 h at 37 °C to get the extract solution of the samples. The extract solutions were then filtered (0.22 μm pore size). L929 cells were resuspended in DMEM supplemented with 10% (v/v) Fetal Bovine Serum at a density of 1.0 × 104 cells per mL and 100 μL of cell resuspension solution was pipetted into 96-well micrometer plates. After incubated at 37 °C under 5% CO2 atmosphere for 24 h, the medium was replaced by the previously prepared extracted dilutions (50 vol%), with the culture medium as blank control and DMSO as negative control. After 24 h, 48 h, and 72 h of incubation, the morphologies of the cells in the plate were observed by using an inverted phase contrast microscope (Olympus IX50-S8F2). The cells were treated with 20 μL per well MTT (5 mg mL−1 in PBS solution) and incubated for another 4 h at 37 °C in a humidified atmosphere of 5% of CO2. Then the culture medium was removed and 200 μL per well of DMSO was added to dissolve the formed formazan crystals. After the plate was shaken for 15 min, the optical density (OD) was read on a multi-well microplate reader at 630 nm. The cytotoxicity for each membrane was tested by six averages of extract substrate. The relative growth rate (RGR%) was calculated according to the formula as below:39
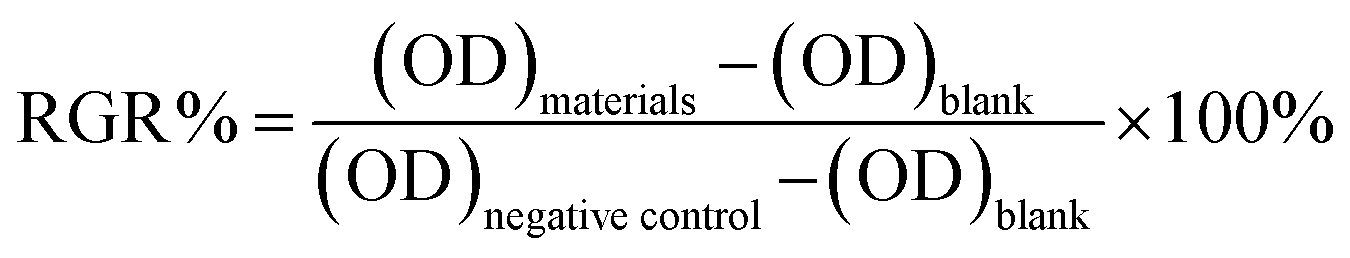 | (3) |
Blood compatibility was evaluated by hemolysis%. Typically, ethylenediaminetetraacetic acid (EDTA)-stabilized human blood samples were freshly obtained from healthy adult volunteers. First, 5 mL of blood sample was added to 10 mL of 0.9% saline, and then red blood cells (RBCs) were isolated from serum by centrifugation at 2000 rpm for 10 min. The RBCs were further washed 3 times with 10 mL of 0.9% saline. The purified RBCs were then diluted to 50 mL 0.9% saline. The PEG–PCL–PLLA-based membranes (10 × 10 mm2 in area) were washed with distilled water 3 times and then put into a test tube with 9.8 mL 0.9% saline and incubated for 30 min at 37 °C. After that, 0.2 mL of diluted blood was added into test tube and incubated for 2 hours at 37 °C. Similarly, 0.2 mL of diluted blood was added to 9.8 mL of distilled water and 0.9% saline solution using as a positive and negative controls, respectively. After the incubation, all the samples were centrifuged at 2000 rpm for 10 min. Then OD values were determined for the absorbance at 540 nm using a spectrophotometer (UV-2550, Japan). The hemolysis percentage (hemolysis%) can be calculated by the following equation:40,41
 | (4) |
Results and discussion
Synthesis of copolymer and structure characterization
To obtain multiblock copolymers consisting of PEG, PCL and PLLA units via urethane linkages, difunctional building blocks were required. For this purpose, double hydroxyl-terminated linear PEG–PCL–PLLA prepolymers with different molecular weight ratios were prepared. Firstly, PEG–PCL diols were synthesized; the structures were confirmed by the appearance of peak f at 4.24 ppm belonging to methylene protons of the PCL–CO–OCH2–CH2–O–PEG segment (Fig. 1(a)). PEG–PCL diols were further used as the macro-initiator to prepare PEG–PCL–PLLA prepolymers. The purified PEG–PCL–PLLA polymers were characterized by 1H NMR (Fig. 1(b)). As shown in Fig. 1(b), the peaks at 5.25 ppm and 4.38 ppm corresponded to the methane proton b and to the methane proton of lactide end group b' respectively. To analyze the crystalline properties systematically, calculated by 1H NMR of the prepolymers, the relative proportion of PEG, PCL and PLLA blocks were obtained, as shown in Table 1. | ||
| Fig. 1 1H NMR of (a) PEG–PCL; (b) PEG–PCL–PLLA; (c) the typical products, namely, sample C with ethoxy terminal group and D with POSS terminal; and (d) FTIR of selected samples. | ||
| Sample | nPEGa | xPCLa | yPLLAa | Mwb | PDIb | T. F. | Contact angle (°) |
|---|---|---|---|---|---|---|---|
| a Calculated from 1H NMR.b Obtained by GPC. T. F. replaced terminal functional group. | |||||||
| A | 1 | 1.52 | 0.48 | 111350 |
1.530 | EtO– | 81.48(0.93) |
| B | 1 | 1.37 | 1.19 | 84200 |
1.458 | EtO– | 87.86(2.45) |
| C | 1 | 1.56 | 1.52 | 93780 |
1.483 | EtO– | 83.12(0.54) |
| D | 1 | 1.56 | 1.52 | 97600 |
1.437 | POSS– | 103.49(2.67) |
| E | 1 | 1.43 | 2.67 | 81330 |
1.414 | EtO– | 83.58(0.96) |
| F | 1 | 0.93 | 1.13 | 99260 |
1.421 | EtO– | 77.86(3.21) |
Starting from the synthesized PEG–PCL–PLLA diols, a polycondensation using HDI was carried out. As shown in Fig. 1(c), the signal at 3.18 ppm was assigned to the methene groups conjoint to the –CONH– group,42 which confirmed the formation of urethane groups. Furthermore, the signal corresponding to the PLLA end groups (peak at 4.38 ppm) disappeared almost completely, also confirming that the condensation reaction proceeded correctly. Some other characteristic peaks were marked in Fig. 1(c). In addition to 1H NMR spectroscopy, FTIR measurements were conducted to confirm the molecular structure of the copolymers. The peak near 1732 cm−1 was the carbonyl group stretching from ester and amide groups, and peaks at 1625 cm−1 and 1534 cm−1 were attributed to amide I and amide II of amide groups in urethane. Additionally, the signals at 0.97 ppm of methyl and 0.62 ppm of methine in POSS (Fig. 1(c)) were assigned to the POSS terminal because sample D was washed two times with excessive hexane to remove any unreacted POSS.
In addition, bismuth ethylhexanoate, an almost nontoxic chemical, could effectively catalyze the chain extension reaction,43 in conjunction with the mentioned four-step strategy to synthesize linear multi-copolymers with well-controlled chain lengths ratio, which provided a realistic possibility for industrialization.
Crystalline properties
It was well known that crystallinity played an important role in the degradation behavior. As it can be seen in XRD patterns, Fig. 2(a), all samples showed very sharp crystalline reflections at 2θ = 21.4° and 23.6°, corresponding to (110) and (200) lattice planes of PCL.26 While, PLLA segments showed sharp crystalline reflections at 2θ = 14.7°, 16.6° and 19.0°, corresponding to (010), (200) and the composite of (014) and (203) lattice planes.44 However, the PEG segments were amorphous and there was no PEG crystalline reflection in sample A based on XRD. The crystallinity of the sample of A, B, C, D, E and F was calculated to be approximately 42.55%, 28.23%, 17.68%, 32.49%, 34.42% and 21.55%, respectively, from the XRD by using JADE. | ||
| Fig. 2 (a) XRD of PEG–PCL–PLLA-based multi-block copolymers obtained by solution casting method, (b) degree of crystallinity of sample A, B, C and E, with PEG:PCL:PLLA composition value of 1:1.52:0.48, 1:1.37:1.19, 1:1.56:1.52 and 1:1.43:2.67, respectively. | ||
Analyzing sample A, B and E, we found that PLLA crystallization ability was enhanced obviously with the increase of PLLA content, which can be affirmed by the increase in the intensity of (200) crystal plane (Fig. 2(a)). On the contrary, (110) crystal plane intensity of PCL was significantly reduced, indicating that crystallization ability of PCL was weakened. It was because PLLA disrupted the crystallization of PCL, resulting in imperfect crystal structures and subsequently decreasing in melting temperature and crystallinity. When PCL was the main components, the crystallization of PLLA was interfered, directly resulting in no diffraction peak in XRD and melting peak in DSC curve for sample A, so PLLA in the sample A was almost amorphous. Thus we concluded that only when PLLA content reached a certain extent, can PCL and PLLA crystal coexist in the copolymer matrixes. When the content of PLLA was further increased, the chemical regularity of the copolymer matrix was further damaged, which gave rise to the crystallinity decreasing (sample B). When the PCL and PLLA components were with similar proportion, its chemical regularity reached the minimum, endowing the lowest degree of crystallinity for sample C. As to sample D, even in the similar component as sample C, the introduction of POSS with strong interaction, which may play the role of molecular nucleation agents, was conducive to the crystallization of polymers, especially to the PCL blocks, resulting in the crystallinity increasing remarkably. Namely, sample D was with extremely higher crystallinity than sample C. As it can be seen from Fig. 2(a), the relative intensity of PCL crystal plane (200) in sample D increased significantly comparing to sample C, which also indicated that the POSS played a role in inducing crystallization. In addition, sample D with POSS-terminal showed significant POSS crystallization peak, as marked in Fig. 2(a), which indicated that POSS in the sample formed crystalline aggregates.
As shown in Fig. 2(b), with the increase of the PLLA content, the degree of crystallinity in total reduced firstly and increased later. When PLLA content increased to the value of sample E, PLLA itself formed a good crystallinity, which contributed to recovery of degree of crystallinity.
Competitive crystallization of the multi-block copolymers
Melting properties of the copolymers were determined by DSC as shown in Fig. 3. The endothermic peaks at temperature close to 50 °C corresponded to the melting of PCL, while those of higher temperature (about 125 °C) corresponded to the melting of PLLA. We observed that PLLA regions exhibited multiple melting peaks except for sample A (Fig. 3(b)), which can be attributed to the presence of crystals with different size.26 It was evident that the Tm as well as the enthalpies of melting was strongly influenced by the composition of the copolymer. As shown in Fig. 3(a), when the first heating temperature reached 180 °C (Process I), polymer segments were all in melting state. During the quench process, the slow crystallization of PLLA resulted in the presence of PLLA imperfect crystals. Accordingly, the cold crystallization of PLLA (∼90 °C) emerged (except for sample A) in the second heating. PCL can exhibit better crystallinity than PLLA, which was due to relative fast crystallization rate of PCL. However, both sample C and F showed dual cold crystallization. It was mostly attributed to uncompleted crystal of PCL and PLLA because the mole ratio of PCL and PLLA equaling to 1:1, which resulted in low chemical regularity of whole chains.
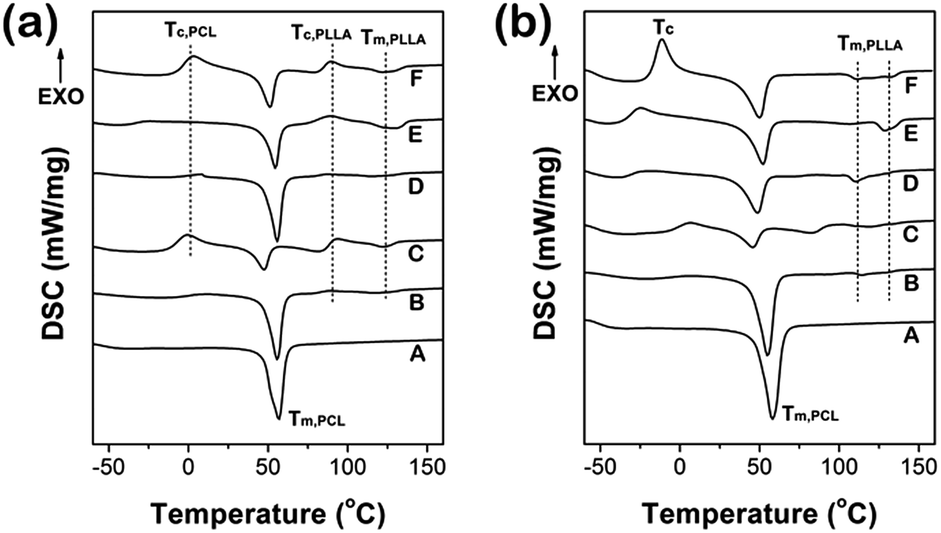 | ||
| Fig. 3 DSC curves of PEG–PCL–PLLA multi-block copolymers with different thermal treatment: (a) heating to 180 °C and then quenched (Process I), (b) heating to 100 °C and then quenched (Process II). | ||
When the first heating temperature reached 100 °C (Process II), as shown in Fig. 3(b), only PCL segments were in melting state, and the PLLA segments were annealing which ensured the PLLA crystallize completely. Therefore, there was no obvious cold crystallization of PLLA during second heating. Furthermore, the crystallization of PLLA had occurred sufficiently in the situation, which resulted in the values of PLLA crystallinity being generally higher than that obtained from Process I except for sample D, as shown in Fig. 4(a).
 | ||
| Fig. 4 Crystallinity of (a) PLLA and (b) PCL segments with different thermal treatment: Process I, heating to 180 °C and then quenched; Process II, heating to 100 °C and then quenched. | ||
The results showed that, when PLLA was with high crystallinity (Process II), crystallinity of PCL in all samples except for sample D were lower than that obtained from Process I, as shown in Fig. 4(b). This indicated that during the heat treatment, the crystallization ability of the components was in a competition state, which was termed “competitive crystallization”. Clearly, PCL and PLLA each can form individual crystalline microstructure due to the different unit cell parameters as well as crystal conformation, giving rise to microphase separation driven by thermodynamic incompatibility; in addition, the PCL and PLLA (even POSS for sample D) were in one molecular chain. Therefore, high crystallinity of one component will significantly restrict the crystallization ability of another component. For typical example, the complete PLLA crystallization of sample F (Process II) resulted in a greater degree of inhibition of PCL crystallization, which can be affirmed by the PCL cold crystallization in Fig. 3(b) being stronger than that in Fig. 3(a).
The crystallization of sample D was different with other samples. On the one hand, appropriate amount of POSS aggregation may induce the crystallization of PCL and PLLA by acting as nucleation agents. Thus, in the Process I, we observed that under the same PCL and PLLA components and heat treatments, the crystallinity of both PCL and PLLA in sample D was significantly higher than that in sample C (Fig. 4). On the other hand, the terminal POSS with strong interaction will restrict the thermal movement so as to crystallization of the whole molecular chain. Accordingly, both PLLA (Fig. 4(a)) and PCL (Fig. 4(b)) in sample D showed lower crystallinity than that obtained in Process I because of restriction of POSS aggregates on the ordered arrangement of PLLA and PCL segments under the heat treatment in Process II. The data based on DSC were all listed in Tables 2 and 3.
| Samples | Tg (°C) | Tc,PCL (°C) | ΔHc,PCL (J g−1) | Tm,PCL (°C) | ΔHm,PCL (J g−1) | Tc,PLLA (°C) | ΔHc,PLLA (J g−1) | Tm,PLLA (°C) | ΔHm,PLLA (J g−1) | φc,PCL% | φc,PLLA% | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Onset | Peak | |||||||||||
| A | −53.4 | — | — | 47.3 | 56.7 | −65.82 | — | — | — | — | 64.64 | — |
| B | −60.1 | — | — | 47.8 | 55.7 | −42.31 | 87.7 | 1.29 | 118.6 | −2.91 | 52.32 | 5.81 |
| C | −54.8 | −2.0 | 11.24 | 39.4 | 47.4 | −20.12 | 91.3 | 4.46 | 122.3 | −4.90 | 11.18 | 1.43 |
| D | −50.3 | — | — | 48.4 | 55.6 | −52.12 | 86.1 | 1.28 | 116.9 | −3.81 | 65.60 | 8.23 |
| E | −55.0 | −26.2 | 2.41 | 47.4 | 54.5 | −35.52 | 88.8 | 5.58 | 129.5 | −11.78 | 54.79 | 13.84 |
| F | −54.3 | 2.7 | 22.49 | 43.5 | 51.2 | −30.4 | 88.7 | 7.33 | 122.2 | −8.12 | 11.66 | 2.42 |
| Samples | Tg (°C) Mid. | Tc,peak (°C) | ΔHc (J g−1) | Tm,PCL (°C) | ΔHm,PCL (J g−1) | Tm,PLLA (°C) | ΔHm,PLLA (J g−1) | φc,PCL% | φc,PLLA% | |
|---|---|---|---|---|---|---|---|---|---|---|
| Onset | Peak | |||||||||
| A | −50.8 | — | — | 49.2 | 58.1 | −57.4 | — | — | 56.37 | — |
| B | −54.8 | — | — | 37.1 | 48.8 | −24.9 | 110.2 | −5.53 | 30.79 | 19.83 |
| C | −52.0 | 5.6 | 6.29 | 35.9 | 45.6 | −12.3 | 120.1 | −3.63 | 7.56 | 11.82 |
| D | −51.2 | — | — | 45.8 | 54.9 | −48.6 | 114.1 | −2.24 | 61.17 | 7.29 |
| E | −54.7 | −25.9 | 9.11 | 41.7 | 52.1 | −28.1 | 107.0, 129.3 | −1.214, −5.584 | 31.42 | 15.18 |
| F | −51.4 | −11.6 | 26.68 | 38.4 | 50.0 | −29.5 | 112.3–131.7 | −7.569 | 4.16 | 23.15 |
Fig. 5 presented polarizing optical micrographs of the multi-block copolymers, which were prepared by different heat treatment. As it can be seen, sample A showed good spherulitic morphology in Fig. 5, which was due to the highest chemical regularity. The typical PCL spherulites were marked in Fig. 5(a) and (b). With the increase of PLLA content, the chemical regularity decreased, thus samples B, C and D all showed smaller spherulites than that of sample A. Furthermore, as discussed above, the crystallinity of PLLA treated in Process I was lower than that treated in Process II due to the high cooling rate as well as slow crystallization rate of PLLA. Therefore, no obvious PLLA spherulites were observed in Fig. 5(c). However, during the Process II, sample C had enough annealing time, giving rise to significant PLLA spherulites (Fig. 5(g)). In addition, because of the competitive crystallization between PCL and PLLA segments in one chain, the sufficient crystallization of PLLA will restrict crystallization of PCL more, which leaded to smaller spherulites in Fig. 5(e)–(h) comparing to that in Fig. 5(a)–(d), respectively. Also according to DSC analysis, in Process I, POSS in sample D induced crystallization of the PCL and PLLA significantly, so sample D presented typical spherulite morphology (Fig. 5(d)); while during Process II, the behavior of the terminal POSS restricted crystallization of PCL and PLLA segments and then restrict formation of typical spherulites of copolymer in Fig. 5(h).
 | ||
| Fig. 5 Polarizing optical micrographs of the multi-block copolymers: (a)–(d) representing sample A, B, C and D, respectively, which were prepared by heating the samples to 180 °C and crystallizing isothermally at 40 °C for 2 h; (e)–(h) representing sample A, B, C and D, respectively, which were prepared by heating the samples to 180 °C, moving directly to another hot plate at 100 °C, and then crystallizing isothermally at 40 °C for 2 h. | ||
Therefore, we concluded that depending on the composition and different heat treatments, samples attained different degree of crystallinity, so as to realize the effective control of mechanical and degradation properties.
Stress–strain curve
The mechanical data were listed in Table 4 and stress–strain curves of the selected samples were shown in Fig. 6(a). The copolymers showed different crystalline characteristics as discussed above, which endowed the tunable mechanical properties, such as Young's modules and yielding strength. As shown in Fig. 6(b), for samples A, B, C, D and F, both Young's modules and yielding strength increased with the increasing of crystallinity. However, when PLLA content reached as sample E, the intrinsic properties of crystalline PLLA would contribute to the mechanical properties, eventually accounting for the largest elastic modulus and relative high yielding strength (Fig. 6(a) inset and Table 4). Also for sample E, the elongation at break decreased to 124.68%, which was far less than the application requirements of blood vessel substitute. In addition, there was no remarkable yielding in sample C with lowest modulus (Fig. 6(a)), because it was with the lowest crystallinity. Finally, it was worth noting that the POSS can induce crystallization in the situation and act as mechanical enhancement agent accordingly, which resulted in sample D being with much higher elastic modulus than that of sample C (Table 4) and maintaining a high elongation at break.| Samples | Yielding strength (kPa) | Stress strength (kPa) | Young's modules (MPa) | Elongation (%) |
|---|---|---|---|---|
| A | 110.17(6.09) | 141.29(7.71) | 1.57(0.11) | 746.04(96.55) |
| B | 59.03(17.39) | 37.99(14.56) | 1.13(0.32) | 783.57(46.73) |
| C | — | 139.63(13.90) | 0.24(0.04) | 944.05(53.02) |
| D | 86.84(2.45) | 82.37(8.96) | 1.21(0.11) | 446.09(74.40) |
| E | 104.76(9.36) | 78.96(10.26) | 1.61(0.17) | 124.68(35.54) |
| F | 57.39(5.68) | 76.51(17.06) | 0.53(0.11) | 598.47(81.57) |
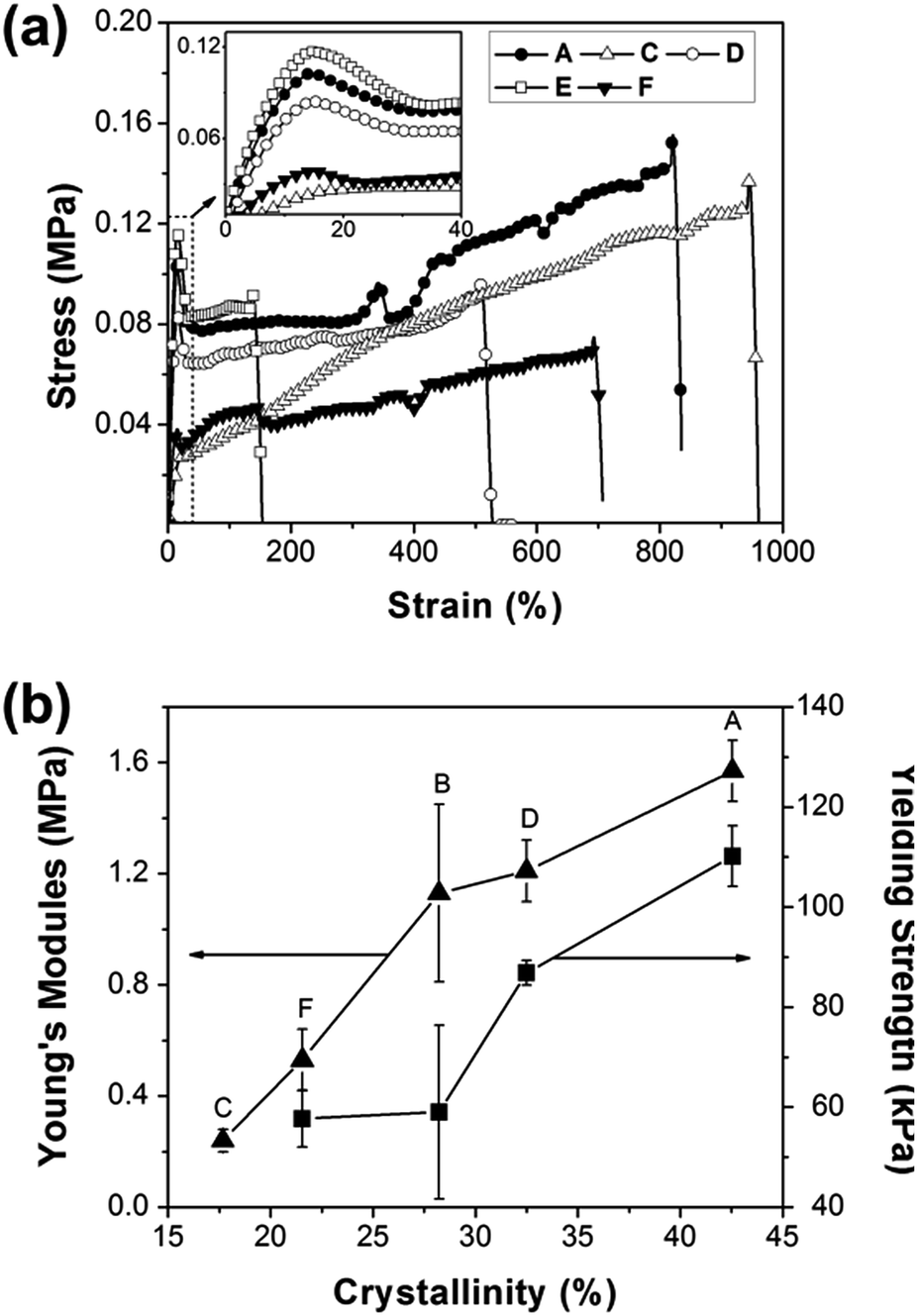 | ||
| Fig. 6 (a) Stress–strain curve of selected samples, (b) curves of Young's modules and yielding strength vs. crystallinity. | ||
Bio-degradation testing
The controllable degradation rate was useful for various biomedical applications45 because it will directly influence the in vivo performance of a scaffold. Changes in composition can vastly change the degradation behavior.33 So it was expected that different weight ratios of PEG, PCL and PLLA in the copolymer might contribute to tunable degradation rates. Consequently, we investigated the effect of composition on the degradation profile of the copolymers by incubating samples in PBS buffer at 37 °C over the course of 2 months.As known, polyesters presented surface or bulk degradation mechanisms depending on the two stages, which was water diffusion and hydrolysis of ester bonds. At the early stage, degradation took place mainly in the surface, so contact angel was necessary to evaluate the surface hydrophilicity. The contact angle for all samples was between 77.86° to 103.49° and listed in Table 1. The key parameters in the bio-degradation process were crystallinity, hydrophilicity and the content of PLLA in the study.
As observed, the high content of PLLA, low crystallinity and high hydrophilicity all accelerated bio-degradation rate. For examples, comparing sample A with sample E (Fig. 7), which were with same PEG–PCL contents, degradation rate of the materials increased significantly with PLLA increasing. Sample B degraded with a rapid rate from the beginning, because it was with low crystallinity; while sample E was with higher crystallinity and giving rise to lower degradation rate accordingly. This was due to the crystalline regions showed the tendency to retard water uptake. Additionally, sample D was with slow degradation rate in 35 days, which could be attributed to the high crystallinity of PCL and the strong hydrophobic effect POSS, giving rise to the highest contact angle with 103.49°. However after 35 days the degradation occurred significantly, herein PLLA ingredient became the dominant factor, resulting in sample D with a faster degradation rate than that of sample A. As to sample F, with the lower PCL as well as highest PEG content, showed the fastest rate of degradation in all mentioned samples. It can be mainly attributed to the increasing of hydrophilicity, namely, with the lowest contact angle of 77.86°, which was conductive to water uptake.
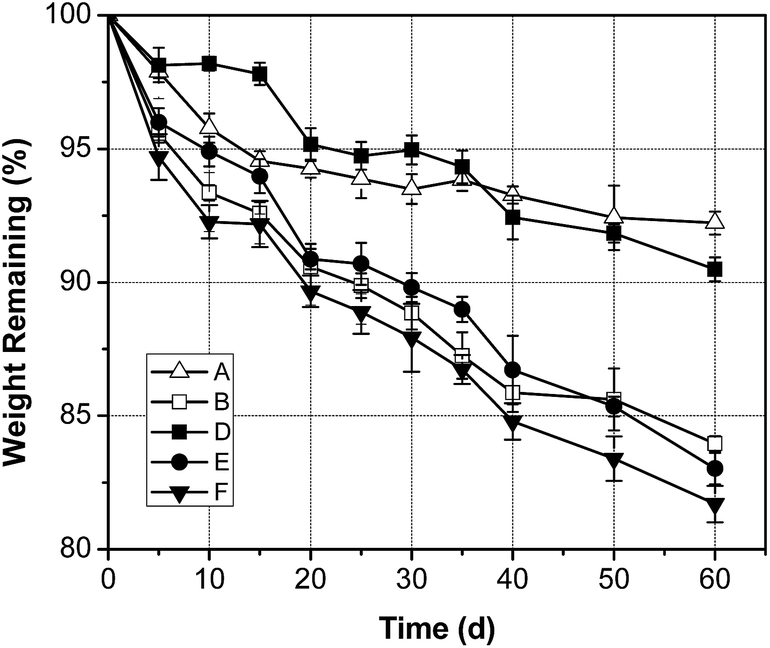 | ||
| Fig. 7 Mass loss profiles of selected samples during 2 months in vitro degradation. The thickness of all selected samples was about 0.4 mm. | ||
Furthermore, the surface morphologies of selected samples were obtained by SEM. As shown in Fig. 8, the roughness of the material surface increased significantly with the occurrence of degradation, which was caused by surface degradation and dissolution. Because the PLLA degraded with a faster rate than that of PCL, the bulges in the surface were considered to be PCL-rich regions, as marked in Fig. 8-a60, e60 and f60 (column 2). The bulk degradation caused the pores (as marked by arrows, column 2) and the cracks (column 3) in the materials, and the crack diffusion eventually resulted in the damage of the material. According to the images in Fig. 8 (column 4), sample A, E and F showed a significant degradation. However, the existence of POSS made sample D exhibit a good step-degradation, namely, the high hydrophobicity of POSS ensure the sample be with a slow degradation rate in the early stage and degraded quickly in the second stage, which can effectively reduce the possibility of thrombosis when it was used for blood treatments. In addition, proposed mechanism of the POSS protection effect was shown in Fig. 9. Concretely, POSS formed aggregates due to the strong interaction, which can act as physical cross-linking points. Under the similar degradation situation, the cross-linking points restricted crack transfer, and connected a plurality of micro-structure unit, ensuring the integrity of sample D after degradation.
 | ||
| Fig. 8 Surface morphologies of selected samples after degradation for 30 days (a30 for sample A, e30 for sample E, f30 for sample F and d30 for sample D, column 1) and 60 days (a60 for sample A, e60 for sample E, f60 for sample F and d60 for sample D, column 2 and 3) obtained by SEM and the images of mentioned samples after bio-degradation for 60 days (column 4). | ||
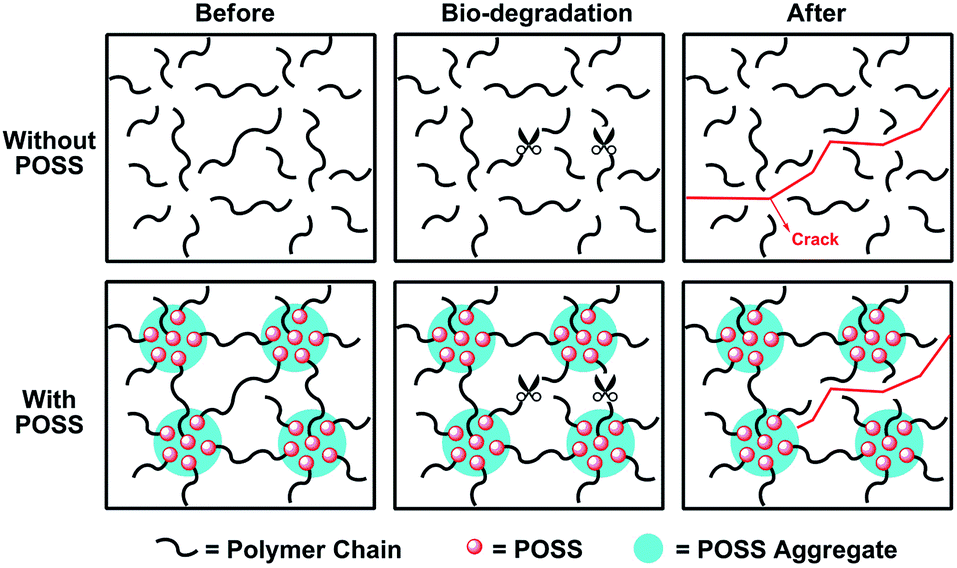 | ||
| Fig. 9 Proposed mechanism of the POSS protection effect on the integrity of materials after degradation. | ||
Thus, we can design the materials with suitable PEG, PCL and PLLA contents to make sure the materials exhibited a controllable degradation rate. In addition, we can introduce POSS to achieve a slow degradation rate in the early stage, and maintain the integrity of the material in a long period. According to the controlled degradation of the project as well as good biocompatibility design principles, we combined the advantages of PEG, PCL, PLLA, and the POSS to prepare PEG–PCL–PLLA–POSS hybrid polymer of biological material having a novel structure and composition. Conveniently, we could achieve different proportions precisely through block copolymerization and “precursor-chain extension” method mentioned in this study.
Cytotoxicity
MTT assay was used to test the cytotoxicity of the membranes. As indicated in Fig. 10, the morphologies of L929 cells incubated for 72 h in the extract substrates of the membranes all showed spindle, triangular, and quadrangular shapes with good growing conditions. The viability of L929 cells cultured in the extract substrates of the membranes had no significant difference with that in the cell culture medium, reflecting that the membranes showed no cytotoxicity. RGR% of sample with POSS termination was highest, which indicated POSS molecules improved the cytotoxicity of materials to some extent. We can find all the RGR% values of the cells cultured on the composite substrates for 3 days were all above 80.77%, which indicated the materials showed no toxicity on the cell viability. | ||
| Fig. 10 Photomicrographs of the L929 cells incubated for 72 h in the negative control and the extract substrates of samples composites (a–f). (a for blank; b for sample A; c for sample B; d for sample D; e for sample E and f for sample F), (g) relative growth rate (RGR%) of L929 cells cultured for 24 h, 48 h and 72 h on samples compared with that on control substrate. | ||
Blood-compatibility
The hemolysis ratio was widely used to evaluate the destructive degree of any implant material to erythrocytes.46 As shown in Fig. 11, the hemolysis% of the samples was all in 1.41–3.75% range, which indicated that the materials were all acceptable for clinical application (being less than 5%). It was worth emphasized that the introduction of POSS slightly improved the blood compatibility. In addition, the increasing of PEG content (sample F) was also conductive to the blood compatibility which was probably due to the improvement of hydrophilicity. | ||
| Fig. 11 (a) Photograph of hemolysis experiment procedure, (b) hemolysis% of PEG–PCL–PLLA-based membranes. | ||
Conclusions
In order to obtain a materials with suitable bio-degradation performance and good biocompatibility, PEG–PCL–PLLA-based poly(ester-urethane)s with different segment ratios were successfully fabricated, which showed good cytocompatibility, and blood-compatibility, namely, with RGR% more than 80.77% as well as hemolysis% lower than 3.75%. Moreover, the crystallization, mechanical behaviour and degradation rate could be effectively modulated by adjusting the terminal functional groups and the segment length of PEG, PCL and PLLA in the synthesis of PEG–PCL–PLLA diol precursor. It was worth noting that the POSS not only acted as crystallization reduce agent and enhanced the mechanical properties accordingly, but also ensured good step degradation of materials. The tunable mechanical behaviour, good cytocompatibility and blood-compatibility, in conjunction with the controllable degradation rate, classified these bio-materials for numerous biomedical applications.Acknowledgements
The authors thank the National Natural Science Foundation of China (No. 51273017), Polymer Chemistry and Physics, Beijing Municipal Education Commission (BMEC, No. XK100100640) for financial support.Notes and references
- M. M. Kose, S. Onbulak, I. I. Yilmaz and A. Sanyal, Macromolecules, 2011, 44(8), 2707–2714 CrossRef CAS.
- R. Wang, W. Chen, F. Meng, R. Cheng, C. Deng, J. Feijen and Z. Zhong, Macromolecules, 2011, 44(15), 6009–6016 CrossRef CAS.
- T. Chung-Kan, K. Chao-Yin, Y. Shu-Rui, Y. Chin-Yu, M. B. Eric, H. Simon, C. I. Ming and C. Ming-Huei, Biomaterials, 2014, 35, 1163–1175 CrossRef PubMed.
- P. Y. Ni, Q. X. Ding, M. Fan, J. F. Liao, Z. Y. Qian, J. C. Luo, X. Q. Li, F. Luo, Z. M. Yang and Y. Q. Wei, Biomaterials, 2014, 35, 236–248 CrossRef CAS PubMed.
- H. X. Dan, V. Cédryck, A. G. Fisher, S. M. Hamlet, Y. Xiao, D. W. Hutmacher and S. Ivanovski, Biomaterials, 2014, 35, 113–122 CrossRef CAS PubMed.
- R. Zheng, H. C. Duan, J. X. Xue, Y. Liu, B. Feng, S. F. Zhao, Y. Q. Zhu, Y. Liu, A. J. He, W. J. Zhang, W. Liu, Y. L. Cao and G. D. Zhou, Biomaterials, 2014, 35, 152–164 CrossRef CAS PubMed.
- A. J. Melchiorri, N. Hibino, T. Yi, Y. U. Lee, T. Sugiura, S. Tara, T. Shinoka, C. Breuer and J. P. Fisher, Biomacromolecules, 2015, 16(2), 437–446 CrossRef CAS PubMed.
- C. Huang, S. Wang, L. Qiu, Q. Ke, W. Zhai and X. Mo, ACS Appl. Mater. Interfaces, 2013, 5(6), 2220–2226 CAS.
- J. Guan and W. R. Wagner, Biomacromolecules, 2005, 6(5), 2833–2842 CrossRef CAS PubMed.
- Y. Zhu, C. Gao, X. Liu and J. Shen, Biomacromolecules, 2002, 3(6), 1312–1319 CrossRef CAS PubMed.
- H. Seyednejad, W. Ji, F. Yang, C. F. van Nostrum, T. Vermonden, J. J. J. P. van den Beucken, W. J. A. Dhert, W. E. Hennink and J. A. Jansen, Biomacromolecules, 2012, 13(11), 3650–3660 CrossRef CAS PubMed.
- S. Lee, S. Cho, M. Kim, G. Jin, U. Jeong and J. Jang, ACS Appl. Mater. Interfaces, 2014, 6, 1082–1091 CAS.
- B. A. Allo, S. G. Lin, K. Mequanint and A. S. Rizkalla, ACS Appl. Mater. Interfaces, 2013, 5, 7574–7583 CAS.
- P. Dinarvand, E. Seyedjafari, A. Shafiee, A. B. Jandaghi, A. Doostmohammadi, M. H. Fathi, S. Farhadian and M. Soleimani, ACS Appl. Mater. Interfaces, 2011, 3, 4518–4524 CAS.
- M. Nelson, R. Diana, M. Albino, F. Susana, A. F. Nuno, N. M. Joao, L. R. Rui and M. N. Nuno, ACS Nano, 2014, 8(8), 8082–8094 CrossRef PubMed.
- Y. B. Kim and G. H. Kim, ACS Comb. Sci., 2015, 17, 87–99 CrossRef CAS PubMed.
- Q. C. Zhang, K. Tan, Y. Zhang, Z. Y. Ye, W. S. Tan and M. D. Lang, Biomacromolecules, 2014, 15, 84–94 CrossRef CAS PubMed.
- J. Lv, L. Chen, Y. Zhu, L. Hou and Y. Liu, ACS Appl. Mater. Interfaces, 2014, 6(7), 4954–4964 CAS.
- J. Qiu, J. Li, G. Wang, L. Zheng, N. Ren, H. Liu, W. Tang, H. Jiang and Y. Wang, ACS Appl. Mater. Interfaces, 2012, 5(2), 344–350 Search PubMed.
- F. K. Kasper, K. Tanahashi, J. P. Fisher and A. G. Mikos, Nat. Protoc., 2009, 4(4), 518–525 CrossRef CAS PubMed.
- R. G. Pauck and B. D. Reddy, Med. Eng. Phys., 2015, 37(1), 7–12 CrossRef CAS PubMed.
- Z. B. Ning, N. Jiang and Z. H. Gan, Polym. Degrad. Stab., 2014, 107, 120–128 CrossRef CAS PubMed.
- I. Navarro-Baena, J. M. Kenny and L. Peponi, Polym. Degrad. Stab., 2014, 108, 140–150 CrossRef CAS PubMed.
- I. Navarro-Baena, A. Marcos-Fernández, A. Fernández-Torres, J. M. Kenny and L. Peponi, RSC Adv., 2014, 4, 8510–8524 RSC.
- X. Deng, S. Zhou, X. Li, J. Zhao and M. Yuan, J. Controlled Release, 2001, 71(2), 165–173 CrossRef CAS.
- B. Alvarado-Tenorio, A. Romo-Uribe and P. T. Mather, Macromolecules, 2011, 44(14), 5682–5692 CrossRef CAS.
- K. Wu, M. Huang, K. Yue, C. Liu, Z. Lin, H. Liu, W. Zhang, C. Hsu, A. Shi, W. Zhang and S. Z. D. Cheng, Macromolecules, 2014, 47(14), 4622–4633 CrossRef CAS.
- W. Zhang, Y. Li, X. Li, X. Dong, X. Yu, C. Wang, C. Wesdemiotis, R. P. Quirk and S. Z. D. Cheng, Macromolecules, 2011, 44(8), 2589–2596 CrossRef CAS.
- W. Wang, Y. Guo and J. U. Otaigbe, Polymer, 2009, 50(24), 5749–5757 CrossRef CAS PubMed.
- P. T. Knight, K. M. Lee, H. Qin and P. T. Mather, Biomacromolecules, 2008, 9(9), 2458–2467 CrossRef CAS PubMed.
- Q. Guo, P. T. Knight, J. Wu and P. T. Mather, Macromolecules, 2010, 43(11), 4991–4999 CrossRef CAS.
- X. Gu, J. Wu and P. T. Mather, Biomacromolecules, 2011, 12(8), 3066–3077 CrossRef CAS PubMed.
- P. T. Knight, K. M. Lee, T. Chung and P. T. Mather, Macromolecules, 2009, 42(17), 6596–6605 CrossRef CAS.
- L. Wang, S. Di, W. Wang, H. Chen, X. Yang, T. Gong and S. Zhou, Macromolecules, 2014, 47(5), 1828–1836 CrossRef CAS.
- M. Zhang, H. Liu, W. Shao, K. Miao and Y. Zhao, Macromolecules, 2013, 46(4), 1325–1336 CrossRef CAS.
- E. A. Jackson, Y. Lee and M. A. Hillmyer, Macromolecules, 2013, 46(4), 1484–1491 CrossRef CAS.
- J. Xue, M. He, H. Liu, Y. Niu, A. Crawford, P. D. Coates, D. Chen, R. Shi and L. Zhang, Biomaterials, 2014, 35(34), 9395–9405 CrossRef CAS PubMed.
- T. Wu, M. Frydrych, K. O. Kelly and B. Chen, Biomacromolecules, 2014, 15(7), 2663–2671 CrossRef CAS PubMed.
- W. Guo, H. Kang, Y. Chen, B. Guo and L. Zhang, ACS Appl. Mater. Interfaces, 2012, 4(8), 4006–4014 CAS.
- Z. X. Meng, W. Zheng, L. Li and Y. F. Zheng, Mater. Sci. Eng., C, 2010, 30(7), 1014–1021 CrossRef CAS PubMed.
- H. Wang, Y. Feng, Z. Fang, W. Yuan and M. Khan, Mater. Sci. Eng., C, 2012, 32(8), 2306–2315 CrossRef CAS PubMed.
- L. Wang, X. Jing, H. Cheng, X. Hu, L. Yang and Y. Huang, Ind. Eng. Chem. Res., 2012, 51(33), 10731–10741 CrossRef CAS.
- B. Xu, L. Li, K. Zhang, P. M. Macdonald, M. A. Winnik, R. Jenkins, D. Bassett, D. Wolf and O. Nuyken, Langmuir, 1997, 13(26), 6896–6902 CrossRef CAS.
- T. Isono, Y. Kondo, I. Otsuka, Y. Nishiyama, R. Borsali, T. Kakuchi and T. Satoh, Macromolecules, 2013, 46(21), 8509–8518 CrossRef CAS.
- C. Yang, H. Wu, J. Sun, H. Hsiao and T. Wang, ACS Appl. Mater. Interfaces, 2013, 5(21), 10985–10994 CAS.
- J. Wang, Y. He, M. F. Maitz, B. Collins, K. Xiong, L. Guo, Y. Yun, G. Wan and N. Huang, Acta Biomater., 2013, 9(10), 8678–8689 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |