
Synthesis and characterization of 18F-labeled hydrazinocurcumin derivatives for tumor imaging†
Abstract
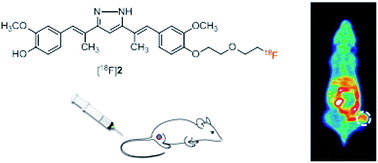
Fluorine-substituted hydrazinocurcumin derivative 1 and its dimethyl-substituted form at the C2 and C6 positions (2) were synthesized and their radiolabeled forms, [18F]1 and [18F]2, were evaluated for tumor imaging. In vitro and in vivo metabolism studies showed that the two radioligands were resistant to reductive metabolism, probably due to the presence of a pyrazole ring. In cellular uptake studies, [18F]1 and [18F]2 exhibited comparable uptake by human umbilical vascular endothelial cells and rat C6 glioma cells. Inhibition of radioligand uptake to a similar extent by HC and curcumin suggests that these radioligands may share the same binding sites as those for HC and curcumin. Positron emission tomography imaging of C6 glioma xenografted mice acquired 30 and 60 min after radioligand injection showed that [18F]2 had markedly higher tumor uptake than [18F]1, which was consistent with biodistribution data (3.20 ± 0.35% ID per g vs. 0.98 ± 0.31% ID per g, respectively). However, the two radioligands showed similar levels of tumor-to-background uptake ratio, except for the significantly higher uptake of [18F]1 by the small intestine, indicating its more rapid clearance. The results of this study will guide further structural modifications of these radioligands to enhance tumor-to-background uptake ratios.
 Please wait while we load your content...
Please wait while we load your content...

