
A self-assembled nanoparticle platform based on poly(ethylene glycol)–diosgenin conjugates for co-delivery of anticancer drugs
Chunxiao Liab,
Lin Daib,
Kefeng Liua,
Lihong Denga,
Tingting Peic and
Jiandu Lei*ab
aMOE Engineering Research Center of Forestry Biomass Materials and Bioenergy, Beijing Forestry University, Beijing, 100083, China. E-mail: ljd2012@bjfu.edu.cn; Fax: +86 10 62337251; Tel: +86 10 62337251
bBeijing Key Laboratory of Lignocellulosic Chemistry, Beijing Forestry University, Beijing, 100083, China
cSchool of Life Science and Technology, Harbin Normal University, Harbin 150025, China
First published on 27th August 2015
Abstract
Diosgenin (DGN) is a steroidal saponin from a therapeutic herb. It is reported to be a kind of potential natural antitumor drug. However, clinical application of diosgenin in cancer therapy is limited due to undesirable pharmaceutical characteristics such as its consequently poor solubility and low bioavailability. Here we developed a nanoparticle platform based on poly(ethylene glycol)–diosgenin (mPEG–DGN) conjugates for co-delivery of anticancer drugs. Firstly, to improve the solubility and bioavailability of DGN, the amphiphilic conjugates mPEG–DGN were made by linking DGN with mPEG. Then they self-assembled stable nanoparticles to deliver another anticancer drug hydroxycamptothecin (HCPT) by a simple nanoprecipitation method. The obtained nanoparticles possessed the appropriate size, high drug loading efficiency of diosgenin and HCPT, slow release of the drugs and high synergistic effects. Hence, the mPEG–DGN nanoparticle is a promising drug delivery system for cancer therapy.
1. Introduction
Cancer is currently a major concern in public health-care systems, as it is one of the main causes of morbidity and mortality worldwide.1 Diosgenin, the aglycon of dioscin, is a spirostanol saponin containing a trisaccharide unit isolated from the tuberous roots of yam. Diosgenin plays a predominant role in the treatment of malignancies, and it is also considered a lead compound in the discovery of novel anticancer drugs and also as a probe for elucidating cancer molecular mechanisms.2 However, like most potent anticancer drugs, DGN has many problems in clinical application such as poor water solubility, rapid blood clearance, relatively short half-life, and severe side effects for healthy tissues.3In addition, evolution of drug resistance in cancer cells has been attributed as a major factor in the failure of many forms of monotherapy.4 To prevent the cancer cell from evolutionary development, it often requires a high drug does to kill the whole cancer cell population, but what followed was the severe side effort.5 The limitations of monotherapy can be circumvented by synergistic combination of multiple anticancer agents which allows for reduction of the drug, therefore preventing or delaying the emergence of drug resistance. Generally, synergistic combination of two or more drugs is a promising strategy to overcome undesirable toxicity and other side effort that limit the utility of many potential drugs by countering biological compensation, allowing reduced dosage of each agent or accessing context-specific multiple targets.6–9
Poly(ethylene glycol) (PEG) is the most widely used in the modification of small molecular or macromolecular drugs because of its simple synthetic steps and high aqueous solubility. Moreover, PEGylation is well-known for its capability to solubilise very insoluble small molecule compounds, prolong circulation time, and alter the biodistribution of parent drugs.
In this study, an amphiphilic polymer–drug conjugate poly(ethylene glycol)–diosgenin (mPEG–DGN) was synthesized based on mPEG and diosgenin, in which diosgenin and mPEG was used as the hydrophobic and the hydrophilic segment, respectively. Then the mPEG–DGN self-assembled into a polymeric nanoparticle with linear mPEG and DGN (Scheme 1). The nanoparticles were developed as an important strategy for drug delivery due to their capabilities of enhancing drug solubility, improving pharmacokinetics and preferentially accumulating in tumor by the enhanced permeability and retention (EPR) effect.10–12 At the same time, the mPEG–DGN NPs served as an ideal carrier of another anticancer drug, HCPT to achieve combination therapy. We further determined the biophysical properties of the nanoparticles including particle size, loading efficiency, in vitro drug release kinetics, in vivo antitumor activities and side effort.
 | ||
| Scheme 1 Synthesis of mPEG–DGN (A). Illustration of mPEG–DGN co-delivery of HCPT (B). | ||
2. Materials and methods
2.1. Materials
Methoxy poly(ethylene glycol) carboxylic acid (mPEG–COOH, Mn = 5000) was purchased from JenKem Technology Co., Ltd (Beijing, China), it is FDA and EU food grade materials. Diosgenin and hydroxycamptothecin was purchased from Chengdu Preferred Biotechnology Co., Ltd (Sichuan, China). 4-Dimethylaminopyridine (DMAP), dimethyl sulfoxide (DMSO) and 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydrochloride (EDC) were supplied by J&K Chemical Reagent Co., Ltd (Beijing, China). Penicillin and streptomycin, Gibco Dulbecco's Phosphate-Buffered Saline (DPBS), Gibco Dulbecco's Modified Eagle's Medium (DMEM) were all bought from Invitrogen HyClone. The Cell-Counting Kit-8 (CCK8) kit was supplied by the Dojindo Laboratories.Murine Lewis lung carcinoma (LLC) cells were obtained from the Peking University Health Science Center (Beijing, China). The cell were grown in the listed medium: DMEM with 10% FBS, 1% streptomycin–penicillin and maintained in an incubator supplied with 5% CO2/90% air humidified atmosphere at 37 °C.
Female C57BL/6 mice, 6–7 weeks age, were purchased from the National Institute for the Control of Pharmaceutical and Biological Products. All animal experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals, and approved by the Experimental Animal Ethics Committee in Beijing.
2.2. Synthesis of mPEG–DGN conjugate
The mPEG–DGN was synthesized as followed. In brief, mPEG–COOH (2.00 g, 0.4 mmol) and diosgenin (1.33 g, 3.2 mmol) were dissolved in 25 mL of dichloromethane. The solution was cooled to 0 °C and then EDC (0.58 g, 1.8 mmol) and DMAP (0.38 g, 3.2 mmol) were added. The mixture was stirred at 0 °C for 1 h then at room temperature overnight. All the procedures were maintained under nitrogen. The solvent was removed under vacuum. Then the residue was dissolved in 10 mL of dichloromethane. The crude product was precipitated into ice diethyl ether (200 mL). Subsequently, the solids were washed twice with diethyl ether (2 × 250 mL) after filtration. Then the mixture was dried under vacuum to give mPEG–DGN and yield as white powder (90% yield). The purity of the mPEG–DGN conjugate was detected by high performance liquid chromatography (HPLC).2.3. Preparation of drug-loaded nanoparticles
The HCPT loaded mPEG–DGN nanoparticles (mPEG–DGN/HCPT NPs) were prepared by a precipitation method.13 In brief, mPEG–DGN conjugate (20 mg) was dissolved in distilled water (3.5 mL) and then HCPT (10 mg) dissolved in dimethyl sulfoxide (DMSO) (0.5 mL) was added dropwise to the mixture under strong stirring. After being stirred for 20 min at 25 °C, the solution was vortexed for 10 min. The mixture were transferred to a MWCO 3500 cartridge, and dialyzed against normal saline (100 mL) for 2 h with exchanges of dialysate.2.4. Characterizations of mPEG–DGN NPs and mPEG–DGN/HCPT NPs
The hydrodynamic size and ζ-potential of the polymeric nanoparticles were measured by a dynamic laser scattering spectrophotometer (Zetasizer Nano-ZS, Malvern Instruments Ltd, Malvern, UK). Two sample solutions of the nanoparticles prepared by the methods mentioned before were used for the measurement (concentration: 0.1 wt%).The morphology of the mPEG–DGN and mPEG–DGN/HCPT NPs were examined using a transmission electron microscope (JEM-100CXa TEM) with an accelerating voltage of 100 kV. A drop of the nanoparticle solutions (20 μL) was deposited onto the surface of Formvar coated copper TEM grids (Ted Pella, Redding, CA) and allowed to air dry at 25 °C before measurement. The 1H nuclear magnetic resonance (NMR) spectra of the free drug DGN and mPEG–DGN were measured in deuterated chloroform (CDCl3) using a 700 MHz NMR spectrometer (Bruker Advance III 700M spectrometer).
2.5. Measurement of drug loading efficiency
The drug loading efficiency (DLE) of HCPT was analyzed by high performance liquid chromatography (HPLC) (Agilent 1200) equipped with a VYDAC 214TP54 column (C18, 300 Å, 5 μm, 4.6 × 250 mm). The HCPT was detected with a UV detector at 266 nm. The sample was prepared by dissolving 1 mg of mPEG–DGN/HCPT NPs in 1 mL acetonitrile and using the calibration curve of HCPT in the same solvent. The sample volume is 20 μL. The mobile phase is acetonitrile/water solution (28/72, v/v) with a flow rate of 1.0 mL min−1. The column temperature is 25 °C. Drug loading efficiency (DLE) was calculated according to the following equation: DLE (%) = (weight of loaded drug/weight of nanoparticles) × 100%.2.6. In vitro drug release assay
The release of HCPT and DGN from nanoparticles was monitored in PBS solution at different pH values 7.4 and 5.5.14 To characterize the drug release, mPEG–DGN/HCPT NPs were introduced to dialysis bag (MWCO 3500 Da). This dialysis bag was introduced into a bottle with 5 mL of PBS (1 mg mL−1) at pH 7.4 or 5.5. At predetermined time, the whole media was taken and replaced with fresh PBS media. The HCPT concentration in the medium was evaluated by high performance liquid chromatography (HPLC). Each stability profile represents the average of three independent runs with the same sampling schedules. The standard deviation of each point is typically 2% or less.2.7. In vitro cytotoxicity assays
The cytotoxicity of free DGN, free HCPT, blank mPEG–DGN NPs and mPEG–DGN/HCPT NPs were determined using the CCK-8 assay. We used Lewis lung carcinoma (LLC) cell lines which were supplied by the Peking University Health Science Center. The LLC cell lines were grown in RPMI 1640 supplemented with 10% FBS, 1% streptomycin–penicillin, and subcultured 3 times per week. The cells were grown in a humidified incubator at 37 °C, 5% CO2. Briefly, 3 × 103 LLC cells per well in 180 μL culture medium were seeded in 96-well plate (corning, USA) and incubated in the humidified incubator for 24 h at 37 °C. The samples of the drugs were prepared as followed. The free DGN, free HCPT, blank mPEG–DGN NPs and mPEG–DGN/HCPT NPs were dissolved in dimethyl sulfoxide (Merck, Darmstadt, Germany) and then diluted in tissue culture medium. Then the cells were treated with various concentrations of those drugs. After that the cells were incubated for 48 h and 72 h at 37 °C with 5% CO2, respectively. After predetermined time, 30 μL of CCK-8 solution was added to each well and then incubated for 1 h at 37 °C avoid light. The CCK-8 assay was performed on an infinite M200 microplate spectrophotometer at 450 nm and 650 nm.The CCK-8 assay was used for cell evaluation of different samples.15,16 IC50 was obtained as concentrations which inhibited the growth of 50% of the LLC cells.17 IC50 was calculated using the Boltzmann sigmoidal function from Origin 8.6 (OriginLab, Northampton, USA). Data are representative of three independent experiments.
We evaluated the synergistic effects between DGN and HCPT in the nanoparticles by applying the combination index (CI) DGNcombined/DGNsingle + HCPTcombined/HCPTsingle, whereby DGNcombined and HCPTcombined represent the IC50 of drugs used in the combination treatment, and DGNsingle and HCPTsingle represent single drug IC50 values. An index lesser than 1 denotes drug synergism, while a value larger than 1 is an antagonistic effect.18
2.8. Confocal laser scanning microscopy (CLSM) observation
The cellular uptake behavior of free HCPT and mPEG–DGN/HCPT NPs was determined by CLSM toward LLC cells. The cells were trypsinized and seeded on the culture slides (BD Falcon, Bedford, MA) at a density of 6.0 × 105 cells per well (surface area of 8 cm2 per well) after LLC cells achieved 70–80% confluency. The cells were incubated for 24 h at 37 °C. Then the original medium was replaced with free HCPT and mPEG–DGN/HCPT NPs (IC50) and incubated for 4 h at 37 °C. After incubation, the cells were washed with PBS (pH 7.4) at 3 times and fixed with 4% formaldehyde for 10 min at room temperature. The liquid content was then dried completely. The cell nuclei were stained by DMEM with DAPI (H-1200; Vector Laboratories, Inc., Burlingame, CA) for 15 min according to the standard protocol provided by the supplier. The fixed cell monolayer was finally washed three times with PBS and visualized under a laser scanning confocal microscope (TCS SP5, Leica).2.9. In vivo antitumor efficiency
Tumor xenograft model was established in the right axillary flank region of female C57BL/6 mice (6–7 weeks) after injection of 3 × 106 LLC cells. Treatments were initiated when tumors reached an average volume of 100 to 150 mm3, and this day was designated as day 0. On day 0, these mice were randomly divided into 5 groups (n = 6) and administered intravenous injection with PBS (control), free DGN (10 mg kg−1), free HCPT (10 mg kg−1), mPEG–DGN/HCPT NPs (10 mg kg−1, HCPT equivalent) every other day until day 8. All groups were intravenous injected via the tail vein. The tumor sizes and body weights were closely monitored every other day to evaluate the antitumor activities and systematic toxicities of various drugs. The tumor volume for each time point was calculated using the following formulation: (length × width2)/2.19,20 For efficacy studies, the percentage of tumor inhibition (% TGI) was calculated using the following formulation: [(C − T)/C] × 100, where C in the mean tumor volume of the control group and T is the mean tumor volume of the treatment group at the same time. After 6 weeks or when the implanted tumor volume reached 5000 mm3, the mice were sacrificed for humane reasons. To evaluate the hematological toxicity of the nanoparticles, 200 μL of blood was sampled from each mouse after final administration to test the while blood cell number (WBC) using a hematology analyzer (MEK-7222K, Nihon Kohden Celltac E).2.10. Side effort
The detection of allergic reaction is very necessary and important because toxic side-effects of the current chemotherapeutical drugs often cause severe reduction in the quality of life. Some natural anti-cancer drugs such as docetaxel, paclitaxel, and teniposide cyclosporine were usually associated with a high incidence of the type I hypersensitivity reaction.21,22 It has been demonstrated that IgE antibody plays an important role in mediating type I hypersensitivity reaction. Thus, we selected IgE levels as the parameter for rapid evaluation of type I hypersensitivity reaction. Herein, five groups of tumor-bearing mice (26–28 g, n = 6) were used in allergy testing studies (control, DGN, HCPT, mPEG–DGN NPs, mPEG–DGN/HCPT NPs). The samples were administrated via tail intravenous injection every two days at the DGN or HCPT does of 10 mg kg−1 body weight. After administration with different samples for 10 days, orbit blood of mice in different groups was collected and centrifuged. Serum sample were analyzed according to the procedure of mouse IgE ELISA.2.11. Statistical analysis
All experiments in this study were performed three times, and the data were expressed as the means ± standard deviation (SD). ANOVA was utilized to determine statistical significance between different groups. In all analysis, p < 0.05 was considered to be statistical significance.3. Results and discussion
3.1. Synthesis of mPEG–DGN conjugate
The amphiphilic mPEG–DGN conjugate was synthesized as illustrated in Scheme 1. The structure of the conjugate was characterized by 1H NMR spectra. As shown in Fig. 1A, the proton of DGN, mPEG segments were detectable in deuterated chloroform (CDCl3). Fig. 1A and B shows the 1H NMR spectra of DGN, mPEG–DGN. The signals at δ 0.70–2.18 are attributed to the most characteristic peak protons of DGN, δ 3.50–3.85 are attributed to the protons of mPEG. The 1H NMR suggested the successful synthesis of mPEG–DGN. | ||
| Fig. 1 1H NMR spectra of DGN in CDCl3 (A), mPEG–DGN in CDCl3 (B). | ||
3.2. Characterization of the nanoparticles
Amphiphilic polymers with hydrophilic and hydrophobic segments can self-assemble into micelles like nanoparticles in aqueous solution. In this study, the amphiphilic conjugate mPEG–DGN self-assembled into nanoparticles in aqueous phase for loading HCPT (Scheme 1). The mPEG–DGN NPs and mPEG–DGN/HCPT NPs were prepared by a nanoprecipitation technique.23 The DLE of the drug-loaded nanoparticles were listed in Table 1. The HCPT loading content is 14 wt%, suggesting the successful loading of HCPT into the mPEG–DGN NPs.| Compound | DLEHCPT (wt%) | Size (nm) | Zeta potential (mV) |
|---|---|---|---|
| mPEG–DGN NPs | — | 122.41 ± 19.68 | −4.17 ± 0.19 |
| mPEG–DGN/HCPT NPs | 13.20 ± 1.18 | 190.1 ± 29.35 | −5.61 ± 0.27 |
The size distributions of the nanoparticles were determined by DLS assay (Fig. 2B and D). The hydrodynamic radius (Rh) of the blank mPEG–DGN NPs and the drug loaded mPEG–DGN/HCPT NPs were listed in Table 1. The size of the particles was increasing because of the loaded drug, while maintain a narrow distribution. The surface zeta potential is a key factor for a better blood compatibility and prolonged circulation time of the nanoparticles for the reduced clearance by the reticuloendothelial system (RES).24,25 The zeta potential of the nanoparticles were determined by a Nano-ZS Zeta Sizer (Malvern Instruments Ltd, Malvern UK) and the results were listed in Table 1.
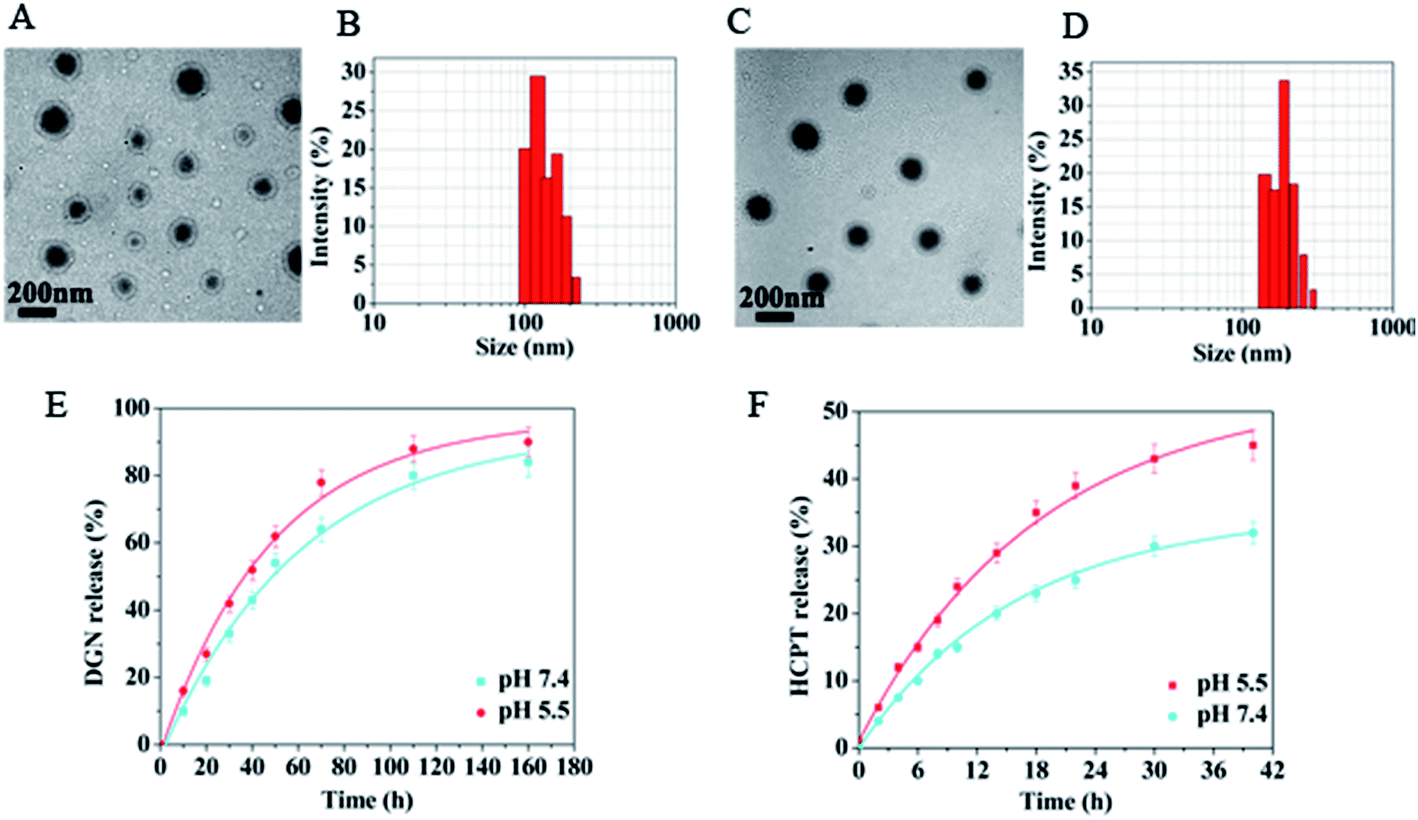 | ||
| Fig. 2 mPEG–DGN NPs (A and B) and mPEG–DGN/HCPT NPs (C and D) observed by TEM (scale bar = 200 nm) and DLS. Drug release kinetics in PBS at 37 °C from the mPEG–DGN/HCPT NPs (E: DGN and F: HCPT). | ||
The morphologies of the blank mPEG–DGN NPs and mPEG–DGN/HCPT NPs were determined by transmission electron microscopy (TEM). The nanoparticles processed uniformly spherical shapes (Fig. 2A and C).
3.3. Release behavior of mPEG–DGN NPs and mPEG–DGN/HCPT NPs
The in vitro drug release kinetics of the mPEG–DGN NPs and mPEG–DGN/HCPT NPs were quantified by HPLC. Nanoparticles with suitable size were first placed in aqueous solutions that simulated biological fluids, and in vitro DGN and HCPT release kinetic were quantified by HPLC analysis using a UV detector at 266 nm (Fig. 2E and F). The mPEG–DGN NPs and mPEG–DGN/HCPT NPs were slowly hydrolyzed and released DGN and HCPT at a weakly acidic or natural pH (5.5 and 7.4) without burst release. This might due to a more effective drug/carrier interaction for mPEG–DGN/HCPT mixed nanoparticles. Moreover, DGN has a benzene ring. In addition to hydrophobic interaction with HCPT, the π–π stacking also contribute to the overall carrier/HCPT interaction.263.4. In vitro cytotoxicity studies
The cytotoxicity studies of the nanoparticles was evaluated using CCK-8 assay. As shown in Fig. 3, free DGN, HCPT, mPEG–DGN and mPEG–DGN/HCPT exhibited potent in vitro cytotoxicity against Lewis lung carcinoma (LLC) cell lines. The IC50 of the various drugs listed in Table 2. DGN exhibited comparable or slightly stronger potency than HCPT with the cell line LLC. The results demonstrated that DGN of nanoparticles is releasable in cancer cells thanks to the ester linker cleavage. After co-delivery with HCPT, nanoparticle mPEG–DGN/HCPT NPs presented a little more potent than mPEG–DGN NPs, confirming the enhanced anticancer efficacy of the co-delivery system. | ||
| Fig. 3 Cellular cytotoxicity of DGN, HCPT, mPEG–DGN NPs, mPEG–DGN/HCPT NPs in LLC cells. CCK-8 assay of DGN, HCPT, and nanoparticles with different concentrations (equivalent to native DGN and HCPT) in LLC cell lines (n = 3, error bars represent standard deviation). | ||
| Compound | LLC (μg mL−1) |
|---|---|
| DGN | 23.4 |
| HCPT | 10.1 |
| mPEG–DGN NPs | 0.48 |
| mPEG–DGN/HCPT NPs | 0.21 |
To compare the potency of the nanoparticles, the concentrations of drug which killed 50% of the cells (IC50) were estimated from the survival curves as shown in Fig. 3 and Table 2. The results showed that the IC50 values of the samples are in the order DGN > mPEG–DGN NPs > HCPT > mPEG–DGN/HCPT NPs (Table 2). The mPEG–DGN/HCPT NPs IC50 value of LLC was 0.21 μg mL−1, the calculated combination index (CI) values of DGN and HCPT in the mPEG–DGN/HCPT NPs were 0.29, which is slightly smaller than 1. This suggested that mPEG–DGN/HCPT NPs achieves a slightly synergistic effect by co-delivery of two different anticancer drugs DGN and HCPT.
3.5. Cellular uptake
The drug accumulation and subcellular localization of nanoparticles were determined by CLSM observation. As shown in Fig. 4, the cellular uptake efficiencies of the free HCPT, mPEG–DGN/HCPT NPs were examined to demonstrate the penetration effects of the nanoparticles into the cells. The internalization of different samples incubated for 4 h were examined. The cellular nuclei were stained with DAPI (blue). In Fig. 4, the fluorescence of HCPT (green) and DAPI (blue) can observed. Nearly no uptake was observed in the HCPT group, as there was no green fluorescence associated with the cells. The drugs then diffuse to the respective target site within the cell and exhibit its action.27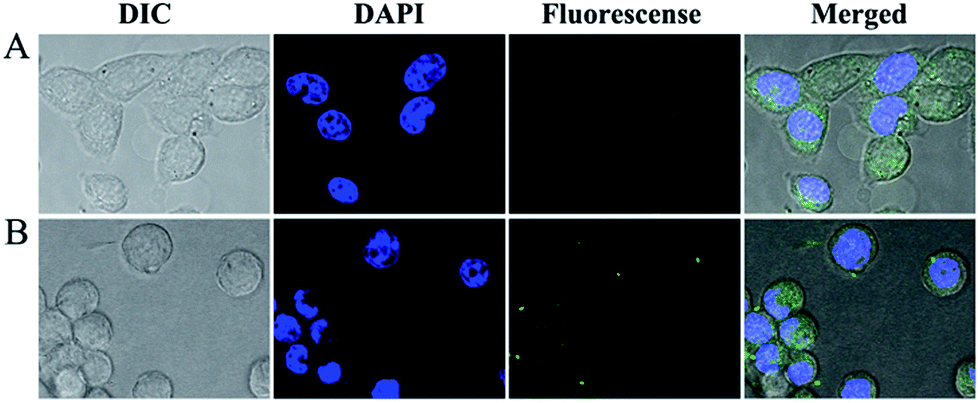 | ||
| Fig. 4 Confocal microscopic pictures of LLC cells incubated with free HCPT (A), mPEG–DGN/HCPT NPs (B) at equivalent HCPT concentration of 0.3 μg mL−1 (IC50) for 4 h at 37 °C. | ||
3.6. In vivo antitumor efficiency
On the basis of the above results, the in vivo antitumor were explored in a mouse tumor model. In vivo antitumor efficiency of drug delivery nanoparticles was compared with efficacy and systemic toxicity of the mPEG–DGN/HCPT NPs free DGN and HCPT at equivalent does of 10 mg kg−1 DGN and 10 mg kg−1 HCPT respectively, on multiple-does schedule in were explored in a mouse tumor model. In vivo antitumor efficiency of drug delivery nanoparticles was compared with xenograft models of lung tumor. As shown in Fig. 5B, tumor-bearing mice treated different drug formulations showed a clear survival advantage compared with the control group treated with PBS. The groups treated with free DGN, HCPT, and virous of nanoparticles showed varied levels of antitumor effects and they were ranked as mPEG–DGN/HCPT NPs > HCPT > mPEG–DGN NPs > DGN. mPEG–DGN/HCPT NPs result in 84.53% TGI (20 day) and 83% survival of animals (30 day). Free DGN result in 36% TGI (20 day) and 0% survival of animals (30 day). Free HCPT resulted in 46.7% TGI (20 day) and 50% survival of animals (30 day) and mPEG–DGN NPs resulted in 78.93% TGI (20 day) and 66% survival of animals (30 day) (Fig. 5A and B and Table 3).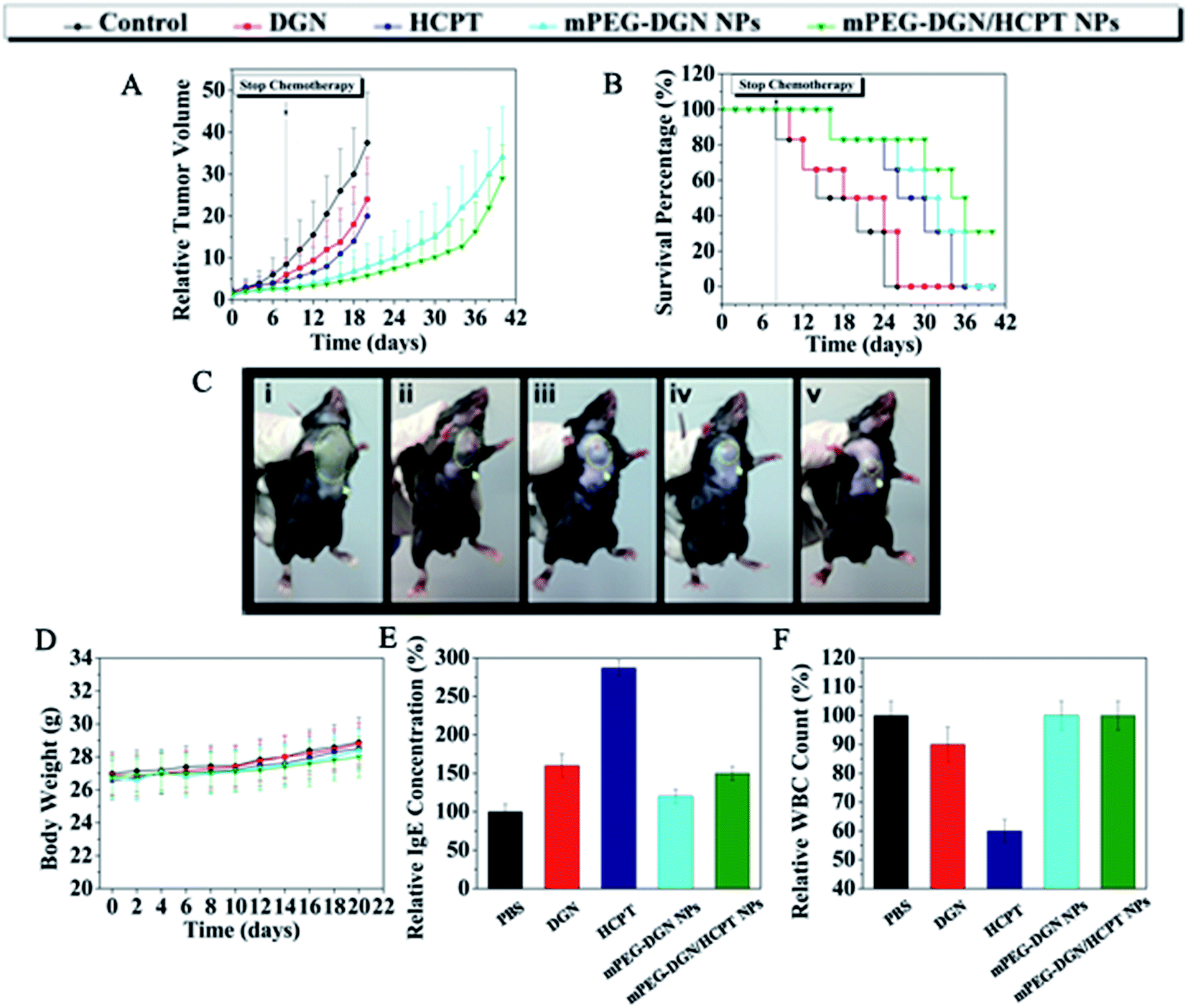 | ||
| Fig. 5 Antitumor efficacy of DGN, HCPT, mPEG–DGN NPs, mPEG–DGN/HCPT NPs in LLC-bearing mice model. Tumor volumes of mice with different drug formulations; dashed line: stop chemotherapy (A). Survival percentage of different treatments (B). Tumor photograph from each treatment groups excised on day 20 ((i): control, (ii): DGN, (iii): HCPT, (iv): mPEG–DGN NPs, (v): mPEG–DGN/HCPT NPs) (C). Body weight variation of mice with different treatments (D). IgE levels of mice treated with different drug formulations for 30 min (E). WBC change during different administrations in normal mice. Blood samples were collected from mice on day 2 after the last treatment. Data are shown as means ± SD; n = 6 (F). | ||
| Compound | Mean TV ± SDa (mm3) | RTVa | TGIa (%) | Curesb (%) |
|---|---|---|---|---|
| a Mean tumor volume (TV), RTV, and % TGI data were taken at day 20 (by day 20, a significant percentage of control animals were euthanized due to excess tumor burden).b % cures were taken at day 30. | ||||
| Control | 4583 ± 1687 | 37.5 ± 12.4 | 0 | 0 |
| DGN | 3041 ± 1269 | 24.1 ± 10.3 | 36.1 | 0 |
| HCPT | 2596 ± 1253 | 19.7 ± 9.6 | 46.7 | 50 |
| mPEG–DGN NPs | 1003 ± 467 | 7.9 ± 5.5 | 78.9 | 66 |
| mPEG–DGN/HCPT NPs | 730 ± 385 | 5.8 ± 3.8 | 84.5 | 83 |
Body weight change is an indicator of systemic toxicity. As shown in Fig. 5D, the body weight of control, free DGN, free HCPT, mPEG–DGN NPs and mPEG–DGN/HCPT NPs treated mice showed a continuous and slow increase. The treatment with the nanoparticles did not lead to any significant body weight loss, demonstrating the reduced systemic toxicity of the free drugs. With high antitumor efficacy and low drug-related toxicity, the mPEG–DGN/HCPT NPs system is promising in cancer therapy.
3.7. Evaluation of the side effort
Many drug formulations, such as paclitaxel and docetaxel are always associated with serious side effort including hematological toxicity and hypersensitivity reactions (usually type I hypersensitivity). Herein, we tested the potential side effort of the nanoparticles. The IgE antibody level and the parameter for rapid evaluation were detected in each group. The blood IgE levels of mice in different groups (PBS, DGN, HCPT, mPEG–DGN NPs, mPEG–DGN/HCPT NPs) are shown in Fig. 5E. The blood IgE of mice which treated with DGN and HCPT displayed higher IgE levels than the PBS group. This might because of the undesirable water solubility. As expected, no significant change of IgE level was observed in mPEG–DGN NPs and mPEG–DGN/HCPT NPs groups, which explored the idea that the use of these nanoparticles could reduce the risk of hypersensitivity reactions substantially. After treated with different drug formulations, the blood of mice was collected to test the WBC count, which is often used as an indicator of hematological toxicity. As Fig. 5F showed, the total WBC count of mice treated with DGN showed a slight decrease over the normal group. No significant decrease in WBC number of the mice treated with mPEG–DGN NPs and mPEG–DGN/HCPT NPs groups were observed, indicating that the nanoparticles could avoid severe hematotoxicity.4. Conclusions
In summary, mPEG–DGN nanoparticles with a hydrophobic core of DGN and a hydrophilic shell of PEG have been designed and synthesized which is capable of encapsulated another anticancer drug HCPT to achieve a synergistic effect, and then their anticancer effects have been evaluated. These nanoparticles for co-delivery of anticancer drugs demonstrated a series of attractive properties as an anticancer drug delivery nanoparticle, including ease of preparation, uniform formulation, appropriate size, high loading capacity of drugs, good stability, high synergistic effects and no side effort.Acknowledgements
This work was supported by the China State Forestry Administration 948 Project (No. 2014-4-35), and National Science Foundation of Beijing, China (Grant No. 2142024), and the National Natural Science Foundation of China (No. 20976179).Notes and references
- A. Jemal, F. Bray, M. M. Center, J. Ferlay, E. Ward and D. Forman, Ca-Cancer J. Clin., 2011, 61, 69–90 CrossRef PubMed.
- J. A. Salvador, J. F. Carvalho, M. A. Neves, S. M. Silvestre, A. J. Leitão, M. M. C. Silva and M. L. S. e Melo, Nat. Prod. Rep., 2013, 30, 324–374 RSC.
- F. Li, R. Goila-Gaur, K. Salzwedel, N. Kilgore, M. Reddick, C. Matallana, A. Castillo, D. Zoumplis, D. Martin and J. Orenstein, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13555–13560 CrossRef CAS PubMed.
- C. Holohan, S. van Schaeybroeck, D. B. Longley and P. G. Johnston, Nat. Rev. Cancer, 2013, 13, 714–726 CrossRef CAS PubMed.
- D. Bovelli, G. Plataniotis, F. Roila and E. G. W. Group, Ann. Oncol., 2010, 21, v277–v282 CrossRef PubMed.
- W. G. Kaelin, Nat. Rev. Cancer, 2005, 5, 689–698 CrossRef CAS PubMed.
- C. T. Keith, A. A. Borisy and B. R. Stockwell, Nat. Rev. Drug Discovery, 2005, 4, 71–78 CrossRef CAS PubMed.
- J. Lehár, A. S. Krueger, W. Avery, A. M. Heilbut, L. M. Johansen, E. R. Price, R. J. Rickles, G. F. Short III, J. E. Staunton and X. Jin, Nat. Biotechnol., 2009, 27, 659–666 CrossRef.
- J. R. Sharom, D. S. Bellows and M. Tyers, Curr. Opin. Chem. Biol., 2004, 8, 81–90 CrossRef CAS PubMed.
- E. K.-H. Chow and D. Ho, Sci. Transl. Med., 2013, 5, 216rv214 Search PubMed.
- A. Khdair, D. Chen, Y. Patil, L. Ma, Q. P. Dou, M. P. Shekhar and J. Panyam, J. Controlled Release, 2010, 141, 137–144 CrossRef CAS PubMed.
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS PubMed.
- M. J. Ernsting, W.-L. Tang, N. MacCallum and S.-D. Li, Bioconjugate Chem., 2011, 22, 2474–2486 CrossRef CAS PubMed.
- M. Ding, N. Song, X. He, J. Li, L. Zhou, H. Tan, Q. Fu and Q. Gu, ACS Nano, 2013, 7, 1918–1928 CrossRef CAS PubMed.
- W. Wei, P.-P. Lv, X.-M. Chen, Z.-G. Yue, Q. Fu, S.-Y. Liu, H. Yue and G.-H. Ma, Biomaterials, 2013, 34, 3912–3923 CrossRef CAS PubMed.
- H. Yue, W. Wei, Z. Yue, B. Wang, N. Luo, Y. Gao, D. Ma, G. Ma and Z. Su, Biomaterials, 2012, 33, 4013–4021 CrossRef CAS PubMed.
- F. Unger, M. Wittmar and T. Kissel, Biomaterials, 2007, 28, 1610–1619 CrossRef CAS PubMed.
- T.-C. Chou, Cancer Res., 2010, 70, 440–446 CrossRef CAS PubMed.
- M. J. Ernsting, M. Murakami, E. Undzys, A. Aman, B. Press and S.-D. Li, J. Controlled Release, 2012, 162, 575–581 CrossRef CAS PubMed.
- K. Kim, J. H. Kim, H. Park, Y.-S. Kim, K. Park, H. Nam, S. Lee, J. H. Park, R.-W. Park and I.-S. Kim, J. Controlled Release, 2010, 146, 219–227 CrossRef CAS PubMed.
- L. Dai, X. Cao, K.-F. Liu, C.-X. Li, G.-F. Zhang, L.-H. Deng, C.-L. Si, J. He and J.-D. Lei, J. Mater. Chem. B, 2015, 3, 3754–3766 RSC.
- L. Dai, D. Li, J. Cheng, J. Liu, L.-H. Deng, L.-Y. Wang, J.-D. Lei and J. He, Polym. Chem., 2014, 5, 5775–5783 RSC.
- L. Dai, L. Wang, L. Deng, J. Liu, J. Lei, D. Li and J. He, Sci. Rep., 2014, 4, 5871, DOI:10.1038/srep05871.
- J.-Z. Du, X.-J. Du, C.-Q. Mao and J. Wang, J. Am. Chem. Soc., 2011, 133, 17560–17563 CrossRef CAS.
- S. Lv, Z. Tang, M. Li, J. Lin, W. Song, H. Liu, Y. Huang, Y. Zhang and X. Chen, Biomaterials, 2014, 35, 6118–6129 CrossRef CAS PubMed.
- X. Zhang, J. Lu, Y. Huang, W. Zhao, Y. Chen, J. Li, X. Gao, R. Venkataramanan, M. Sun and D. B. Stolz, Bioconjugate Chem., 2013, 24, 464–472 CrossRef CAS PubMed.
- T. Ramasamy, J. H. Kim, J. Y. Choi, T. H. Tran, H.-G. Choi, C. S. Yong and J. O. Kim, J. Mater. Chem. B, 2014, 2, 6324–6333 RSC.
| This journal is © The Royal Society of Chemistry 2015 |