
In situ incorporation of a S, N doped carbon/sulfur composite for lithium sulfur batteries
Abstract
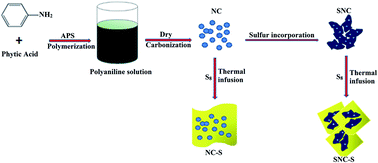
A novel sulfur–nitrogen co-doped carbon material (SNC), which is obtained by taking polyaniline as the nitrogen-containing carbon precursor and then incorporating sulfur atoms in situ as the matrix material for lithium sulfur batteries, is investigated. XPS reveals the formation of strong chemical bonding (–C–S–C– and C–SOx–C (x = 2–4)) in the structure of SNC materials. Moreover, the results demonstrate that SNC–S composites showed higher specific capacity and better cycle performance than the pristine nitrogen co-doped carbon material (NC)/sulfur (NC–S) composites. Except for providing more lithium storage sites due to faradaic reactions, the incorporation of sulfur not only immobilizes the sulfur and polysulfide species, thus improving the interfacial characterization between the electrode and the electrolyte, but also influences the reversible dissolving balance of long-chain polysulfides and suppresses the “shuttle effect”, resulting in better electrochemical behavior. This facile approach of the SNC matrix could provide a practical research direction for lithium sulfur batteries.
 Please wait while we load your content...
Please wait while we load your content...

