
Graphene oxide chemically decorated with Ag–Ru/chitosan nanoparticles: fabrication, electrode processing and immunosensing properties†
Murugan Veerapandian and
Suresh Neethirajan*
Bionano Laboratory, School of Engineering, University of Guelph, Guelph, ON N1G 2W1, Canada. E-mail: sneethir@uoguelph.ca; Fax: +1-519-836-0227; Tel: +1-519-824-4120
First published on 21st August 2015
Abstract
Nanosheets of graphene oxide were chemically decorated with hybrid nanoparticles of a silver–ruthenium bipyridine complex (Ag@[Ru(bpy)3]2+) core and chitosan shell. The oxygenated groups of graphene oxide and the abundant amine groups in the chitosan layer on the surface of the hybrid nanoparticles allow functionalization reactions to occur. Changes in the optical, chemical and structural properties of graphene oxide occurred because the hybrid nanoparticles were studied using spectroscopy and microscopic techniques. Electrodes modified with hybrid nanoparticles-graphene oxide (HNPs-GO) displayed amplified steady-state anodic (IpA) and cathodic (IpC) peak currents with correlation coefficients of 0.9987 (IpA1), 0.9952 (IpA2) and 0.9964 (IpC1). Using the monoclonal antibody Listeria monocytogenes, a HNPs-GO immunosensor could specifically detect L. monocytogenes contaminated in buffer and milk, with a concentration range from 102 to 107 cells per mL and detection limit of 2 cells per mL. Our results suggest that selective optimization of the bio-recognition elements on the HNPs-GO electrode may find prospective use in food processing industries.
1 Introduction
The development of hybrid nanomaterials with multiple structures and chemical composition has attracted many researchers toward the advancement of functional properties.1 Recently, two-dimensional graphene oxide (GO) and reduced GO (rGO) have been used in a variety of applications because of their cost-effective fabrication, ultra-thin layers, large surface area and tunable oxygen functional groups.2,3 Surface treatment and functionalization of the active components on GO nanosheets influence the inherent sp2/sp3 carbon domains, resulting in changing the crystallite size, lattice orientation and associated properties.3,4 The different strategies employed to tune the physico-chemical and biomedical functionalities of GO are photoirradiation, elemental doping and chemical anchoring of inorganic/organic materials.3–6Significant effort has been devoted toward the advancement of hierarchical hybrid GO nanostructures. In particular, ternary/quaternary nanocomposites composed of a graphene derivative, metal, metal oxide and polymer has recently been shown to have improved physico-chemical properties that are optimal for device construction (e.g., electrode materials for biosensor platform and energy conversion). The typical enhanced electrochemical properties of metalloid polymer hybrids (Ag@SiO2-PEG)–GO,5 graphene/WO3/Au7 and polyaniline–Fe2O3–rGO8 are better suited to biosensor studies compared to the individual pristine derivatives. Molecularly imprinted polymers based on CdTe/Cds and magnetic GO showed selective recognition toward environmental pollutants.9 Pt–graphene–TiO2 (ref. 10) and reduced GO–bismuth ferrite (Bi2Fe4O9)11 have been reported to have better photocatalytic properties. Furthermore, studies show that the hierarchical structures of SnS2-rGO–TiO2/TiO2 layered films12 and rGO/Fe3O4@SiO2@polyaniline13 significantly improve the photoelectric and electrochemical properties, respectively. The accumulation of evidence indicates that the fabrication of hybrid GO material has great potential for opto/electrochemical device development.
Nevertheless, achieving a durable structure of hybrid GO with inbuilt multi-functionality is complicated. Physically linked hybrid nanostructures are prone to leaching and decreased synergistic functionality. Compared to the physical adsorption of nanostructures on the GO surface, chemically bonded active materials are expected to have durable electrochemical properties and be more stable.5,6 However, only few studies have demonstrated the chemical functionality between the active materials and GO surface. Incorporation of a durable single hybrid nanostructure on the GO surface with optical, electrochemical and biocompatible capabilities would be highly useful for various biosensing applications. Previous study has shown that single hybrid core–shell nanoparticles made of a metal–dye complex (AgNPs@[Ru(bpy)3]2+) core and biopolymer (chitosan) shell can influence the optical, electrochemical and biocompatibility due to the electrical conductivity of Ag, metal-to-ligand charge-transfer of [Ru(bpy)3]2+ and abundant amino groups of chitosan.14 The three-in-one hybrid14 nanosystem (average particle size 54 nm) on the surface of GO would be an ideal candidate for modification; it is multifunctional due to its opto-electronic and biocompatible nature. GO chemically decorated with hybrid nanoparticles (HNPs) is expected to have better optical, redox activity and biocompatible functional groups suitable for various sensor studies. For example, the introduction of Ag metal on GO improves the electron transfer process and increases the immunosensing ability.15 The presence of metallic and hydrated Ru on the surface of GO electrodes enhances electrochemical performance.16,17 As a bio-derived linear polysaccharide with biocompatibility, biodegradability and film-forming ability, chitosan has been explored as an interface layer in the fabrication of chemically modified electrodes for biosensing.18,19
Chemical functionalization of the distinct materials on the surface of GO is highly dependent on the reactivity of the oxygen functional groups that exist on the edges and basal planes of GO. Herein, for the first time, the three-in-one HNPs composed of Ag@[Ru(bpy)3]2+/chitosan are used to chemically decorate GO nanosheets. Abundant amino groups of the chitosan coating on the surface of Ag@[Ru(bpy)3]2+ provide a significant modification of the oxygenated edges/basal planes of GO. The influence of optical absorbance, photoluminescence, zeta potential and structural integrities, including morphology, chemical structure and Raman shift of pristine GO and HNPs-GO materials, were extensively studied to understand the properties. The inherent electrochemical redox properties were studied on the customized electrode modified with HNPs-GO.
As a proof-of-concept, the electrochemical immunosensing properties of the HNPs-GO electrodes have been demonstrated for the detection of Listeria monocytogenes (Lm). Lm is an important Gram-positive rod-shaped food borne bacterium that causes an extremely life threatening infection, listeriosis (high mortality rate ∼ 30%).20 Listeriosis is most prevalent in pregnant women and their newborns, older adults >65 years old and people with weakened immune systems. A recent report by the CDC (Centers for Disease Control and Prevention), USA (dated June 2015) declared a multi-state outbreak of listeriosis linked to food poisoning.21 The government of Canada estimates that there are about 4 million cases of food borne illness every year. Common symptoms associated with food poisoning include fever, vomiting, diarrhoea and headache. In severe cases, meningitis, septicaemia and abortion can occur.20 Therefore, a rapid and sensitive detection of Lm in food products is vital. Conventional cell cultures, microscopy, biochemical tests and luminescence methods are labor-intensive and time-consuming. Although enzyme-linked immunosorbent assay (ELISA) and polymerase chain reaction (PCR) may greatly reduce the assay time, they are still time-consuming for the identification of the target pathogens and lack the ability of real-time detection.22 Electrochemical based immunosensors are among the most promising because of their high specificity, rapid analysis and field deployable possibility.20,22–24 Herein, electrochemical immunosensing of Lm is proposed using an immobilized mouse monoclonal anti-Listeria monocytogenes antibody (anti-Lm) on the surface of HNPs-GO electrodes. Phosphate buffered saline and milk samples were artificially contaminated with Lm cells and utilized for sensing studies. The cross-reactivity of the proposed immunosensor to another bacterial strain (Pseudomonas aeruginosa, Pa) was also measured and found to be insignificant. In addition to the synergistic electrochemical properties, the biocompatible groups derived from HNPs-GO promote an affinity toward the immobilized antibody, thereby enabling a sensitive detection of Lm contamination.
2 Experimental section
Chemicals
Silver nitrate (AgNO3), 3-mercaptopropionic acid (3-MPA), sodium borohydride (NaBH4), tris(2,2′-bipyridyl)dichlororuthenium(II) hexahydrate, chitosan (low molecular weight: 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–190000 g mol−1; degree of deacetylation: 75–85%), graphite powder (<20 μm, synthetic), glutaraldehyde solution and phosphate buffered saline (PBS) were purchased from Sigma-Aldrich. Mouse monoclonal IgG antibodies to Listeria monocytogenes were purchased from AbD Serotec (Bio-Rad). Milk samples were obtained from a local grocery store. Other chemicals were of analytical grade and used as received without any further purification. Milli-Q water (18.2 MΩ cm) was used for all experiments.
000–190000 g mol−1; degree of deacetylation: 75–85%), graphite powder (<20 μm, synthetic), glutaraldehyde solution and phosphate buffered saline (PBS) were purchased from Sigma-Aldrich. Mouse monoclonal IgG antibodies to Listeria monocytogenes were purchased from AbD Serotec (Bio-Rad). Milk samples were obtained from a local grocery store. Other chemicals were of analytical grade and used as received without any further purification. Milli-Q water (18.2 MΩ cm) was used for all experiments.
Synthesis of the hybrid (Ag@[Ru(bpy)3]2+/chitosan) NPs
Hybrid NPs composed of Ag@[Ru(bpy)3]2+/chitosan were prepared according to the reported procedure.15 At first, 5 mL of AgNO3 (0.1 M), 25 mL of 14 N aq. NH4OH and 5 mL of 3-MPA (50 mM) were dissolved in 15 mL of deionized (DI) water (solution A). Separately, 5 mL of NaBH4 (0.02 M) and 2 mL of 14 N aq. NH4OH were dissolved in 15 mL of DI water (solution B). At room temperature, the solutions of vial A and B were slowly injected dropwise into 300 mL of DI water over 30 min with magnetic stirring at 600 rpm. After 30 min of reaction time, the colloidal solution containing the AgNPs-modified 3-MPA was separated by centrifugation (13000 rpm for 1 h). The particles were then washed twice with DI water and dispersed in DI water for further reaction.
The [Ru(bpy)3]2+ coating on the AgNPs was achieved by mixing the abovementioned AgNPs-modified 3-MPA (5 mL, 1 mg mL−1) and an ethanolic solution of Ru(bpy)3Cl2 (5 mL, 0.8 mg mL−1). The reaction mixture was left overnight under mild stirring, protected from light. The resulting particles were centrifuged (13000 rpm for 1 h) and washed twice with ethanol and DI water to remove the unreacted [Ru(bpy)3]2+. The prepared Ag@[Ru(bpy)3]2+ were then surface modified with chitosan using a coordination chemical reaction between Ag@[Ru(bpy)3]2+ (5 mL, 1 mg mL−1) and chitosan (5 mL, 0.01 wt%) under magnetic stirring of 600 rpm for 3 h at room temperature. The final hybrid (Ag@[Ru(bpy)3]2+/chitosan) NPs were isolated by centrifugation (13000 rpm for 1 h), washed and re-dispersed in DI water for further experimentation.
Functionalization of HNPs on GO nanosheets
Colloidal dispersions of the GO nanosheets used in the current experiment were synthesized according to the modified Hummers method.25 Functionalization of the HNPs onto the surface of GO sheets was achieved by a one-step process. An aqueous dispersion of GO (25 mL, 0.5 mg mL−1) and HNPs (25 mL, 2 mg mL−1) was added to a reaction flask and kept under magnetic stirring (600 rpm) at room temperature for 12 h. After the reaction time has elapsed, the HNPs-functionalized GO sheets were separated by centrifugation (13000 rpm, 1 h), washed thrice with DI water and utilized for characterization.
Construction of HNPs-GO sheets modified electrode
An integrated gold printed circuit board (Au-PCB) chip served as an electrode system. The central circle-shaped Au substrate with an area of 2 mm in diameter was used for the modification of HNPs-GO sheets. Two crescent-shaped Au substrates with a length of 4.3 mm and a breadth of 0.8 mm were used as the counter and reference electrodes, respectively. After performing sequential washing with acetone, ethanol and DI water, the Au-PCB chip was exposed to plasma treatment. Then, typically 4 μL of the aqueous dispersion of HNPs-GO (1 mg mL−1) was drop-casted on the working substrate. To achieve uniform surface modification of HNPs-GO sheets on the electrode surface, typically three layers of casting were performed at regular intervals with an evaporation period of 1 h at ambient temperature.Fabrication of the anti-Lm modified HNPs-GO electrodes
HNPs-GO electrodes were first modified with 4 μL of 1.25% (vol/vol) PBS solution of glutaraldehyde and kept at ambient temperature for 30 min to allow the reaction between the amine groups on the chitosan layer of the HNPs-GO, resulting in an aldehyde functional group at the surface. The terminal aldehyde surface was then coupled to diluted anti-Lm (4 μL, 100 μg mL−1 in PBS, pH 7.4) and kept at 4 °C for 1 h. After this, the modified electrode surface was gently immersed into a PBS buffer (pH 7.4) for 30 s to allow the diffusion of any unbound antibodies away from the electrode surface.Preparation of the bacterial cells and amperometric detection
Bacterial cell suspensions (Lm and Pa) were prepared from overnight cultures, grown in 4 mL of tryptone soy broth culture medium (TSB, Oxoid Canada, Nepean, Ontario). The bacterial cultures were then isolated and washed twice in DI water via centrifugation at 4500 rpm for 10 min at room temperature. Afterwards, the bacterial cells were re-suspended in PBS for further analysis. An optical density measurement at 600 nm was performed for the stock bacterial suspension, and the desired test samples of various cell numbers were diluted appropriately. Chronoamperometric detection of the bacterial samples on the surface of HNPs-GO/anti-Lm electrodes was measured at an applied potential of +0.55 V. Typically, 20 μL of test samples (contaminated PBS and milk) were utilized onto the electrode surface.Instrumentation
A Cary 5000 UV-Vis-NIR spectrophotometer (Agilent Technologies) was used to analyze the UV-vis absorbance spectra. Morphological characterizations were observed using transmission electron microscopy (TEM) (Philips Tecnai 12) with an acceleration voltage of 120 kV. Samples used for imaging were prepared by casting 4 μL of (0.25 mg mL−1) HNPs, GO or HNPs-GO suspension onto a carbon-coated nickel grid. The zeta potential was studied using a Zetasizer Nano ZS (Malvern Instruments) equipped with a 4 mW, 633 nm He–Ne laser using appropriate cells. Measurements were obtained in backscattering (173°) mode and detected with an Avalanche photodiode. For accurate determination of the zeta potential, 13 runs were averaged for each liquid sample. A Varian Cary Eclipse Fluorescence spectrophotometer was used to examine the photoluminescence properties of the HNPs, GO and HNPs-GO. The chemical structure and functional group modifications on the pristine and hybrid materials were identified using Fourier transform infrared (FTIR) spectra obtained using a Nicolet 6700 FTIR spectrometer (in the ATR mode, diamond crystal). 1H-NMR spectra in deuterated dimethyl sulfoxide-d6 (DMSO-d6) were obtained on a Bruker AV 400 spectrometer operating at 400 MHz (number of scans: 256). Raman spectral analysis was performed on a RENISHAW inVia Raman microscope equipped with CCD camera and a Leica microscope. An aqueous dispersion of the sample (∼1 mg mL−1) was drop-casted on a cleaned silica wafer and utilized for measurements. An excitation wavelength of 514 nm and laser power of 10% was used. A short working distance 50× objective lens was used to focus the laser spot on the sample surface. Measurements were carried out in 30 s of exposure time at varying numbers of accumulation. The electrochemical properties of the pristine GO and HNPs-GO materials were studied using cyclic voltammetry on a SP-150 potentiostat and Bio-Logic instruments. All the cyclic voltammograms (CVs) were obtained using a 10 mM PBS solution (pH 7.4) as the supporting electrolyte in the potential region between −0.25 and +0.8 V. A reproducible voltammogram can be obtained under steady-state conditions after about five cycles.3 Results and discussion
Synthesis of the Ag@[Ru(bpy)3]2+/chitosan NPs and functionalization on GO
Fig. 1 illustrates the step-wise synthetic route for obtaining HNPs. The HNPs are composed of a metal–dye complex (Ag@[Ru(bpy)3]2+) core and a chitosan shell firmly bonded to each other via electrostatic and coordination interactions, respectively.14 The thin layer of chitosan on the surface of the HNPs with abundant amine groups are reactive to the oxygenated functional groups of GO. The presence of carboxyl and epoxy groups at the edges and basal planes of GO provide multiple binding sites for chemical functionalization of the HNPs. The two important surface chemical reactions involved in this functionalization were amide formation at the carboxyl groups and nucleophilic attack at the α-carbon by the HNPs. The structural and chemical changes, which resulted from the functionalization process, were characterized using FT-IR and 1H-NMR spectroscopy as described later. | ||
| Fig. 1 Illustration of the stages in the fabrication of HNP (sequential wet-chemical synthesis of AgNPs stabilized with 3-MPA, [Ru(bpy)3]2+ dye complex coating on AgNPs and chitosan modification on Ag@[Ru(bpy)3]2+ via in situ reduction, electrostatic and coordination reaction, respectively) and covalent functionalization on GO nanosheets. | ||
UV-vis absorbance and photoluminescence
In general, the optical absorption and emission band of metal or metal hybrid nanostructures depend on the size, shape, nature of the surface functional layer and solvent environment.26 Herein, UV-vis absorbance spectroscopy was utilized to measure the optical information of the prepared materials. Fig. 2A represents the spectra observed from aqueous AgNPs and AgNPs-modified with 3-MPA. The peak at 402 nm denotes the existence of the characteristic surface plasmon resonance (SPR) of AgNPs. SPR is the collective oscillations of the conductive electrons that exist on the surface of the metal NPs. Depending on the excitation of the localized surface plasmon, caused by strong light scattering at a specific wavelength, strong SPR bands are produced.27 At 423 nm, a significant red shift was mediated by the surface modification of AgNPs with 3-MPA. The UV-vis absorption spectrum of [Ru(bpy)3]2+ shows three specific peaks at 242, 290 and 450 nm (Fig. 2B) ascribed to the intra-ligand π → π* transition, bpy π → π1* transition and metal-to-ligand charge-transfer (MLCT) bands, respectively.28 A shoulder peak at 420 nm was also attributed to MLCT (t2g (Ru) → π* (bpy) transitions). Similarly, AgNPs modified with [Ru(bpy)3]2+ also exhibit three absorbance peaks with a moderate hump located at 423 nm due to the overlap of the SPR from the AgNPs.29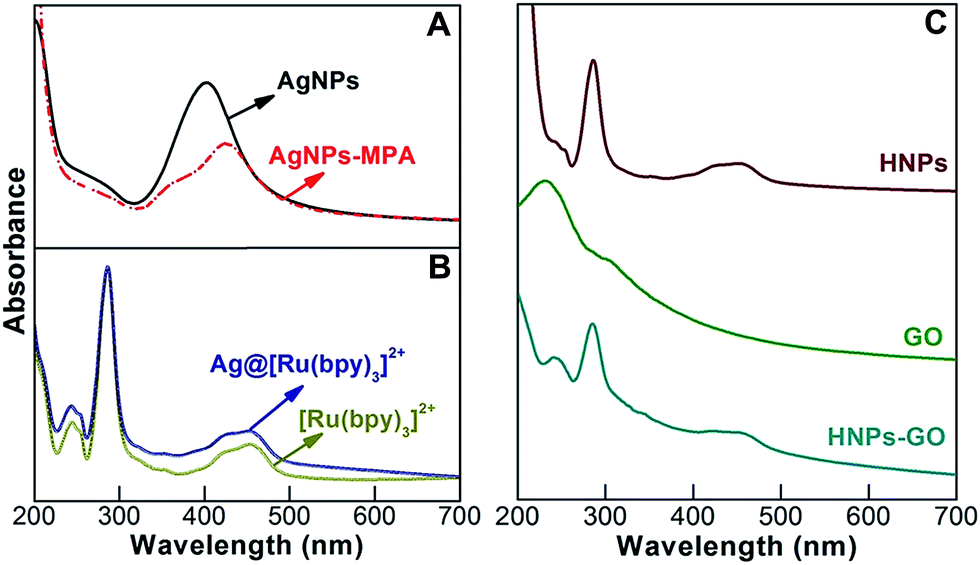 | ||
| Fig. 2 UV-vis absorbance spectra of an aqueous dispersion of (A) AgNPs and AgNPs stabilized with 3-MPA, (B) [Ru(bpy)3]2+ and Ag@[Ru(bpy)3]2+, and (C) HNPs, GO and HNPs-GO. | ||
As shown in Fig. 2C, the HNPs exhibited significant changes in the peak shape at 242, 290 and 450 nm, indicating that the chitosan modification altered the optical absorbance of the Ag@[Ru(bpy)3]2+.14 An aqueous dispersion of GO nanosheets exhibited a wavelength of maximum absorbance at 230 nm, attributed to the π → π* electron transition of the polyaromatic C–C bonds of the GO layers.5 The UV-vis absorbance spectrum of HNPs functionalized GO exhibits peaks centered at 240, 284 and 450 nm. When compared to pristine HNPs, the π → π* electron transition signals of HNPs-GO are well resolved, probably due to the associated signals of the metal–dye complex and C–C bonds of GO. There is no significant change observed from the bpy π → π1* transition peak position; however, the peak centered at 450 nm was much more broader than that observed for the pristine HNPs.
Excitation at specific wavelengths of light on the aqueous dispersion of optically active nanomaterials can provide additional information such as photoluminescence (PL). It is known that the chemical oxidation of graphite results in the formation of mixed sp2/sp3 domains in the GO lattice, which creates a disruption of the π-networks and generates an emission band.30 The PL spectra of GO and HNPs-GO were obtained using an excitation wavelength of 325 nm and are shown in Fig. 3. GO shows a sharp emission peak in the near UV region at around 365 nm due to the amorphous sp3 matrix that surrounds the various graphitic sp2 domains, which act as a high tunnel barrier resulting in the generation of a band gap in GO. This is in agreement with previous studies on the PL of GO.30,31 Upon modification with HNPs, the near band emission was quenched with a slight broadening of the peak centered at 362 nm. This observation was probably due to the formation of new metallic hybrid clusters on the GO lattice. The observation of PL from the HNPs-GO implies the existence of a band gap in the electronic structure of the material. Recent studies have identified that surface modification of GO can create a large band gap and decent carrier mobility suitable for advanced PL32 and electrochemical biosensors.33 The as-prepared chemically decorated GO containing the ternary composite of metal–dye complex and biopolymer retained the inherent PL properties and electronic structure, and is expected to be a feasible option for dual (optical/electrochemical) sensors.
 | ||
| Fig. 3 PL spectra of aqueous GO and HNPs-GO nanostructures (λex = 325 nm) measured at room temperature. | ||
Morphology and surface zeta potential characterization
The morphology of the HNPs, GO and HNPs-GO nanostructures were visualized using TEM, as shown in Fig. 4. HNPs with an overall spherical shape and chitosan coating are clearly visible in Fig. 4A and B. Observed traces of particle aggregation is possibly because of the drying process being performed before imaging. The average particle size distribution of the HNPs was determined using a Malvern dynamic light scattering Zetasizer Nano ZS instrument and found to be 54 nm (data not shown). The surface topography of GO (Fig. 4C and D) displays a corrugated thin sheet-like membranous layer. The typical thin grooves or wrinkles on the sheets are characteristic of GO nanostructures. Due to its two-dimensional thin layered feature with reactive oxygenated groups, GO allows multiple chemical bonding sites with the amine-functionalized HNPs. The HNPs were well decorated on the surface of the GO nanosheets (Fig. 4E and F).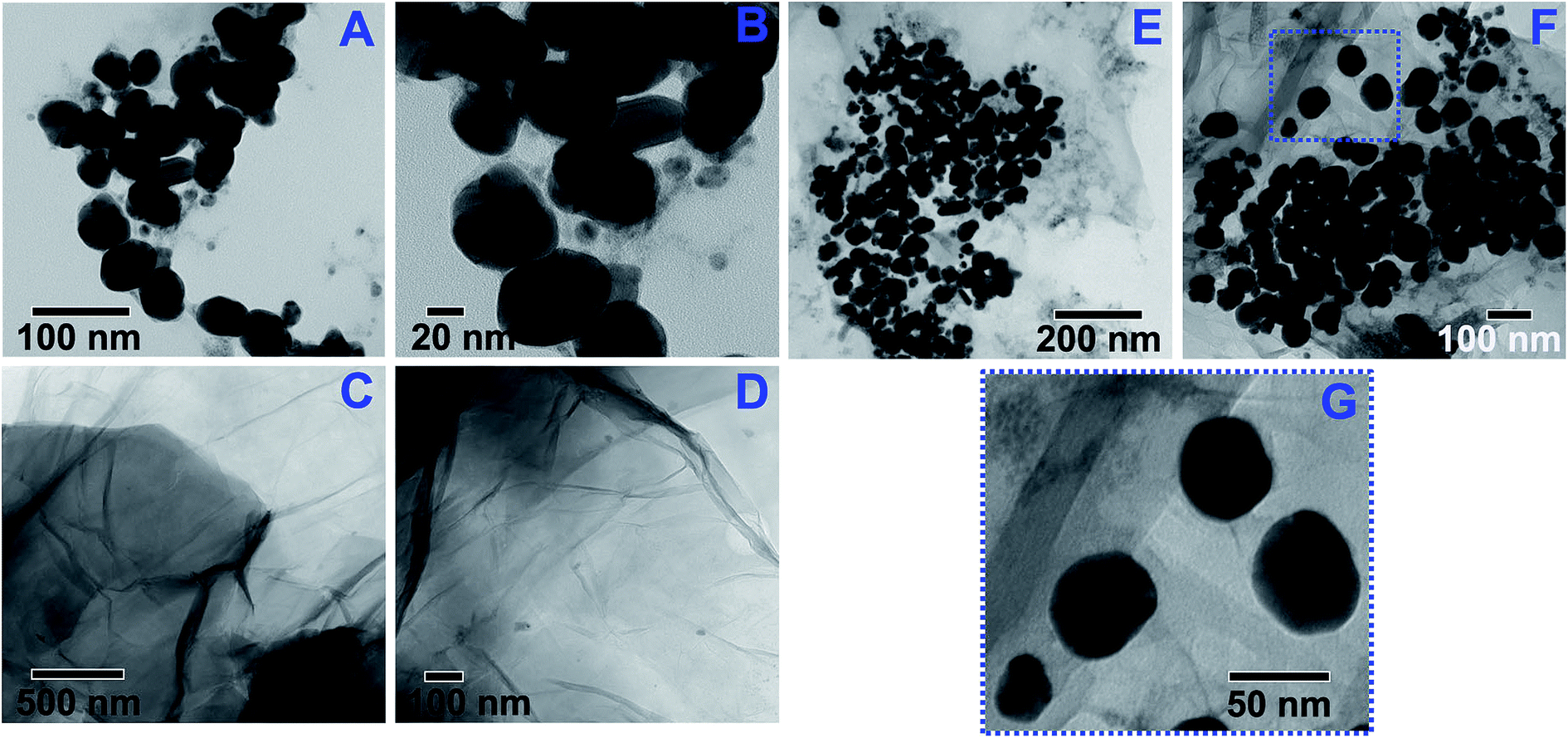 | ||
| Fig. 4 TEM images of the (A and B) HNPs, (C and D) GO and (E–G) HNPs-GO nanostructures. | ||
Zeta potential is a vital physical property used to study the stability of colloidal dispersions and the surface charge associated with the double layer around the colloidal particles. The zeta potential varies depending on which chemical groups exist on the surface of the colloidal particles (ESI Fig. S1†). Due to the ionization of the multiple surface oxygenated functional groups, pristine GO showed a negative zeta potential of −39 mV. HNPs containing a chitosan shell with abundant amine groups displayed the positive zeta potential of +46.1 mV. Upon surface functionalization, the zeta potential of the HNPs-GO was +26.6 mV, indicating that the chemically bonded HNPs modified the inherent surface zeta potential of the GO nanosheets. These results supplement the morphological images. Such modified hierarchical GO sheets with a single hybrid of metal–dye complex and biopolymer provide a unique set of physico-chemical properties that are promising for multi-functional materials.
FTIR and 1H-NMR spectroscopy
In order to evaluate the chemical structure and functional group modifications on the HNPs or HNPs-GO, a comparative FTIR spectral analysis was performed on pristine chitosan and GO samples. Fig. 5A shows the FTIR spectrum of chitosan: the C–H out-of-plane bending at 887 cm−1 and C–O stretching are located at 1026 and 1065 cm−1, whereas C–O–C stretching and C–H bending are located at 1154 and 1377 cm−1. Vibrations at 1590 and 2870 cm−1 are attributed to N–H bending and C–H stretching, and the broad peak centered at 3317 cm−1 was associated with the N–H stretching and hydrogen-bonded OH groups.34,35 The HNPs containing chitosan modified Ag@[Ru(bpy)3]2+ (Fig. 5B) exhibit significant alterations in their group frequencies. For instance, the C–O stretching band shows distinct changes at 1040 cm−1 when compared with pristine chitosan. The primary N–H bending at 1590 cm−1 was shifted to 1644 cm−1, denoting the formation of a secondary amine. A short but sharp peak at 2970 cm−1 was ascribed to the asymmetric stretching of C–H.34 Furthermore, the N–H stretching and H-bonded OH stretching were much more intense than that obtained for chitosan. The observed modifications in the group frequencies (C–O stretching and N–H bending) of chitosan support their chemical bonding with Ag@[Ru(bpy)3]2+.14 | ||
| Fig. 5 FTIR spectra of the (A) pristine chitosan powder, (B) HNPs, (C) GO and (D) HNPs-GO. | ||
The FTIR spectrum of the GO samples (Fig. 5C) reveals the peaks relating to C–O (carbonyl) at 1040 cm−1, the C–O–C epoxy group frequencies at 1175–1250 cm−1 and carboxyl-associated OH signal at 1405 cm−1.13,36 Well-resolved peaks at 1600 and 1725 cm−1 are assigned to the C–C vibrations of the un-oxidized graphitic domains and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations, respectively.5,36 The relatively broad peak centered at 3240 cm−1 was associated with the adsorbed water on the surface of the GO. As discussed previously, HNPs are expected to form chemical bonds at the basal planes and edges of GO. After functionalization with HNPs, the carbonyl peak of GO was broadened and shifted from 1040 cm−1 (Fig. 5C) to 1068 cm−1 (Fig. 5D). The epoxy group frequencies were almost dispersed and the peaks of carboxyl-associated OH, C–C graphitic domains and CO stretching vibrations were also changed. Furthermore, a new peak centered at 771 cm−1, along with two shoulder peaks (at 735 and 830 cm−1), which can be attributed to the C–H bonds of chitosan,14 were observed. This provides evidence of the functionalization of HNPs on the GO surface.
O stretching vibrations, respectively.5,36 The relatively broad peak centered at 3240 cm−1 was associated with the adsorbed water on the surface of the GO. As discussed previously, HNPs are expected to form chemical bonds at the basal planes and edges of GO. After functionalization with HNPs, the carbonyl peak of GO was broadened and shifted from 1040 cm−1 (Fig. 5C) to 1068 cm−1 (Fig. 5D). The epoxy group frequencies were almost dispersed and the peaks of carboxyl-associated OH, C–C graphitic domains and CO stretching vibrations were also changed. Furthermore, a new peak centered at 771 cm−1, along with two shoulder peaks (at 735 and 830 cm−1), which can be attributed to the C–H bonds of chitosan,14 were observed. This provides evidence of the functionalization of HNPs on the GO surface.
To gain further understanding of the chemical structure of the pristine and hybrid nanostructures, 1H-NMR spectroscopy was utilized. Fig. 6A shows the 1H-NMR spectrum of the HNPs, which exhibits the characteristic resonance peaks that can be attributed to the functional groups of chitosan such as –CH–CH– (0.8 and 1.2 ppm), –NH– (1.9 ppm) and OH (5.3 ppm).37 Proton signals of the GO nanostructures (Fig. 6B) are located at 1.2, 4.5, 8.1 and 9.5 ppm and are attributed to –CH–CH–, OH, –C–COOH– and C–COOH, respectively.37,38 The spectrum of HNPs-GO (Fig. 6C) also shows the inherent –CH–CH– protons. The peak shift located at 1.9–2.0 ppm, attributed to the amine protons of chitosan, was relatively weaker than that observed for the pristine HNPs (Fig. 6A), indicating the functionalization of HNPs on GO. The successful chemical bonding of HNPs on the oxygenated functional groups of GO was validated by the absence of free carboxyl proton signals (at 8.1 and 9.5 ppm) and the appearance of multiple amide proton signals (at 6.5, 6.9, 7.1 and 7.2 ppm). The presence of reactive epoxy and carboxyl groups on the GO lattice structures offered the necessary binding sites for the chemical decoration of the HNPs.
 | ||
| Fig. 6 1H-NMR spectra of the (A) HNPs, (B) GO and (C) HNPs-GO samples in DMSO-d6. | ||
Raman spectroscopy
Raman spectroscopy further revealed the structural integrity of GO after chemical interaction with the HNPs. The typical characteristics of the Raman spectra of graphite materials are a G-band at 1570 cm−1, which can be attributed to the E2g phonon of the sp2 C domains,39 and a D-band at 1345 cm−1, which can be attributed to the vibrations of the disordered C domains of graphite.36,39 The presence of a D-band at 1355 cm−1 and a G-band at 1596 cm−1 supports the oxygenation of graphite (Fig. 7). Chemically decorated HNPs on the surface of the GO lattice displayed a broadened D-band at 1355 cm−1 and G-band at 1583 cm−1 (red shifted from inherent 1596 cm−1). A slight change in the intensity ratio of the D- and G-bands (I(D)/I(G)) of HNPs-GO (0.85) compared to that observed for GO (0.81) indicated that the functionalization of the HNPs altered the in-plane sp2 graphitic domains of GO. According to an empirical formula, known as the Tuinstra–Koenig relation,40 the average crystallite size of the ordered graphitic sp2 C domains can be calculated using the following equation,| La (nm) = [(2.4 × 10−10)(λl)4]/[I(D)/I(G)] |
 | ||
| Fig. 7 Raman spectra of GO and HNPs-GO. | ||
Fabrication and redox properties of the HNPs-GO electrodes
Compared to conventional electrodes, carbon electrodes modified with conductive hierarchical nanostructures exhibit an enhanced electron transfer rate and more durable electrochemical properties.7,33 Immobilization of bio-friendly conductive nanostructures with active chemical groups suitable for anchoring an antibody or enzyme is certainly valuable for the fabrication of label-free biosensor platforms.43 To understand its feasibility as a transducer material for an electrochemical biosensor platform, the primitive electrochemical response of the HNPs-GO was evaluated in comparison with pristine GO. Fig. 8 shows the pictorial representation of an integrated three-electrode system used for the modification of pristine GO and HNPs-GO. Unlike conventional electrochemical systems, there is no external counter or reference electrode utilized in the present study. In order to find an optimal potential region suitable for the prepared materials, a pre-screening CV measurement was performed between −1.0 and +1.0 V. From the analysis, it was found that −0.25 to +0.8 V is an optimal potential region for studying the redox behavior of the HNPs-GO modified Au-PCB electrodes. | ||
| Fig. 8 A schematic illustration of an integrated three-electrode system and modification with GO and HNPs-GO nanosheets. | ||
Fig. 9A shows the CV curves for the bare Au-PCB, pristine GO and HNPs-GO modified Au-PCB electrodes, which were obtained at a constant scan rate of 50 mV s−1. A 10 mM PBS solution containing the final concentration of 0.0027 M potassium chloride and 0.137 M sodium chloride at pH 7.4 was used as the supporting electrolyte. Under experimental conditions, the bare Au-PCB and pristine GO electrodes do not exhibit any significant redox behavior. Pristine HNPs without GO as an interface layer are poorly stable on the Au-PCB substrate, which results in leaching and hinders the durable electrochemical response (data not shown). On the other hand, the HNPs-GO modified electrode showed well-defined and highly amplified anodic peaks A1 at +0.38 V and A2 at +0.52 V; the former was related to the oxidation of Ag → Ag2O (ref. 44) and the latter was derived from the oxidation reaction of [Ru(bpy)3]2+ → [Ru(bpy)3]3+. Interestingly, the HNPs-GO electrode displayed a single cathodic peak at −0.12 V, suggesting a coherent reduction reaction of Ag2O → Ag and [Ru(bpy)3]3+ → [Ru(bpy)3]2+. The anodic peak current (A2: Ip = +43 μA) generated from the oxidation reaction of [Ru(bpy)3]2+ was higher than the peak current of A1 (Ip = +33 μA). This was attributed to the existence of [Ru(bpy)3]2+ on the surface of the Ag core nanostructures, which support the overall metal-to-ligand charge transfer process. Furthermore, it was speculated that the presence of the chitosan layer on the core Ag@[Ru(bpy)3]2+ was expected to have a reasonable influence on the observed redox wave.
 | ||
| Fig. 9 (A) CVs of the bare Au-PCB, pristine GO and HNPs-GO modified electrodes. (B) CVs of the HNPs-GO electrode at different scan rates (50–120 mV s−1) in 10 mM PBS (pH 7.4) and (C) the corresponding plots of the anodic (A1 and A2) and cathodic (C1) peak currents against the square root of the scan rate. | ||
Earlier studies demonstrated that the insulating nature of GO nanosheets can be transformed by functionalization of metallic composites, which not only provides better electrical conductivity but also creates a 3D hierarchical environment with a large surface area for rapid electron transfer. For instance, conductive polyaniline interconnected Fe2O3–rGO composites exhibit a surface-confined redox transition at the electrode interface.8 Enhanced redox waves generated from Ag-doped with organometallic or conductive polymer composite electrodes have been reported previously in the study.44,45 Similarly, herein the hybrid combination of the chemically interacted core Ag@[Ru(bpy)3]2+ and shell chitosan enabled a significant redox reaction at the GO interface. Such a hybrid nanoplatform is useful for constructing advanced biosensor platforms.
In order to evaluate the consistency of the redox potentials and increasing peak currents with respect to the scan rate, CVs for the HNPs-GO electrodes were recorded at different scan rates from 50 to 120 mV s−1 (Fig. 9B). The enhancement of the anodic (A1 and A2) and cathodic (C1) peak currents are in relation to the scan rate (Fig. 9C). The correlation co-efficients for the anodic peaks were 0.9987 (IpA1) and 0.9952 (IpA2), whereas that for the cathodic peak was 0.9964 (IpC1), indicating that it was a surface-confined process. This interesting redox behavior, which has emerged from the current investigation on CVs of HNPs-GO, is valuable and provides the possibility of exploring their bio-affinity toward novel molecules through a label-free, direct electrochemical detection strategy.
Immunosensing properties of the HNPs-GO/anti-Lm electrodes
Using the chronoamperometric technique the electrochemical immunosensing properties of the HNPs-GO/anti-Lm electrodes was carried out as a function of Lm concentration. To show the detection ability, the desired concentrations of Lm cells were diluted in PBS and in milk samples, respectively, for the individual amperometric measurements. In general, the antibodies immobilized onto the electrode surface form a layer that would interfere with the inherent electron transfer process at the electrode interface.46 Studies have reported that the membranes of biological cells show a resistance of 102 to 105 Ω cm2.46,47 The adhesion of bacterial cell membranes to the electrode interface, through a specific antibody–antigen complex, can create a further barrier for the electrochemical process. The decrease in the magnitude of the current is related to the number of bacterial cells captured by the immobilized antibodies. Herein, the change in amperometric current was obtained using the experimental group, with Lm and control group, without Lm (ΔI = ILm − Icontrol). Based on the broad and amplified anodic properties of the HNPs-GO electrodes in PBS buffer, an optimal peak potential of +0.55 V, responsible for oxidation of [Ru(bpy)3]2+ → [Ru(bpy)3]3+, has been applied to measure the change in amperometric response. As summarized in Fig. 10A, the amperometric current response from the electrodes shows a linear relationship for the Lm concentration in the range of 102 to 107 cells per mL. The regression equation for the experimental data was y = A + B(X), where y is the sensor response to a change in current (−ΔI), X is the Lm concentrations in cells per mL, and A and B are sensor constants. Linear fitting of the PBS data gave y = −5.43 × 10−6 + 1.82 × 10−5(X) with a correlation co-efficient of 0.98. Likewise a good fit (R2 = 0.98) was obtained for the milk samples: y = 2.55 × 10−5 + 5.61 × 10−6(X). From these correlations, the sensitivity of the HNPs-GO based immunosensor in PBS was calculated to be 1.82 × 10−5 A/101 Lm cells per mL and in milk was 5.61 × 10−6 A/101 Lm cells per mL. Compared to PBS, the sensitivity of HNPs-GO electrodes in the milk samples are decreased perhaps due to the milk components such as fat, cholesterol, carbohydrates and vitamins. The change in the current values to the change in concentration of Listeria Monocytogenes cells in both the milk and PBS samples is linear. The observed sensitivity trend in the PBS and milk samples is in agreement with earlier immunodetection studies.24,48 The detection limit has been determined using the 3σ/m criteria, where σ is the standard deviation of blank and m is the slope of the calibration plot and was found to be 2 cells per mL for both PBS and milk diluted with Lm. The observed lowest detection limit was better than that observed in recent reports.22–24,48 The immunosensing characteristics of the proposed HNPs-GO/anti-Lm electrode have been summarized in Table S1.† Fig. 10B shows the comparative absolute amperometric histogram with different species, Pa. The results demonstrated that, due to the presence of monoclonal anti-Lm, the HNPs-GO-based immunosensor is specific for Lm, thereby interactions with other pathogens do not exhibit a cross-reaction on the sensor surface, resulting in an uninterrupted electrochemical process. The detection approach proposed herein has been shown to be fast in response time, <1 min. This enhanced immunosensing property could be ascribed to the synergistic material's composition of the electrode surface. | ||
| Fig. 10 (A) Calibration curve fit illustrating the sensor response for various concentrations of Lm cells in PBS and milk. The sensor response was expressed as the change in current obtained by the experimental group, with Lm and control group, without Lm (ΔI = ILm − Icontrol). (B) Absolute amperometric current response of the HNPs-GO/anti-Lm electrode, without and with bacterial cells (Lm and Pa) in PBS. The measurements were performed at an applied potential of +0.55 V. Each data point represents the average of three independent measurements at different electrodes and the error bars denote the standard deviation of the mean. | ||
4 Conclusion
The fabrication of GO nanosheets chemically decorated with HNPs composed of a metal–dye complex (Ag@[Ru(bpy)3]2+) core and biopolymer (chitosan) shell was demonstrated using a scalable wet-chemical approach. Functionalized HNPs-GO facilitates a profound improvement in the electron-transfer process at the electrode interface compared to pristine Au, HNPs and GO. The HNPs-GO based electrochemical immunosensor shows a linear correlation between the change in current and various concentrations of L. monocytogenes in PBS and milk samples. The monoclonal anti-Lm modified HNPs-GO electrode showed the lowest detection limit of 2 cells per mL. A selectivity study with P. aeruginosa demonstrated that the proposed immunosensor exhibits no cross-reactivity. The HNPs-GO immunosensing system demonstrated advantages for its ease in fabrication and operation, sensitivity, selectivity and rapid analysis toward the model food pathogen, L. monocytogenes. Additional studies are warranted to understand the impedance changes caused by the interaction of the antibody–bacteria complex on HNPs-GO electrode and its related bacterial number, which will further optimize the sensitivity of the system.Acknowledgements
The authors sincerely thank the Natural Sciences and Engineering Research Council of Canada (Grant# 400929), the Ontario Ministry of Research and Innovation (Grant# 051455) and the Dairy Farmers of Canada (Grant# 051520) for funding this study.References
- K. T. Nguyen and Y. Zhao, Nanoscale, 2014, 6, 6245–6266 RSC.
- F. Bonaccorso, L. Colombo, G. Yu, M. Stoller, V. Tozzini, A. C. Ferrari, R. S. Ruoff and V. Pellegrini, Science, 2015, 347, 1246501 CrossRef PubMed.
- Y. Matsumoto, M. Koinuma, S. Y. Kim, Y. Watanabe, T. Taniguchi, K. Hatakeyama, H. Tateishi and S. Ida, ACS Appl. Mater. Interfaces, 2010, 2, 3461–3466 CAS.
- H. Wang, T. Maiyalagan and X. Wang, ACS Catal., 2012, 2, 781–794 CrossRef CAS.
- M. Veerapandian, Y.-T. Seo, K. S. Yun and M.-H. Lee, Biosens. Bioelectron., 2014, 58, 200–204 CrossRef CAS PubMed.
- M. Veerapandian, N. Lévaray, M.-H. Lee, S. Giasson and X. X. Zhu, ACS Appl. Mater. Interfaces, 2015, 7, 14552–14556 CAS.
- A. Devadoss, P. Sudhagar, S. Das, S. Y. Lee, C. Terashima, K. Nakata, A. Fujishima, W. Choi, Y. S. Kang and U. Paik, ACS Appl. Mater. Interfaces, 2014, 6, 4864–4871 CAS.
- S. Radhakrishnan, K. Krishnamoorthy, C. Sekar, J. Wilson and S. J. Kim, Chem. Eng. J., 2015, 259, 594–602 CrossRef CAS PubMed.
- S. Han, X. Li, Y. Wang and S. Chen, Chem. Eng. J., 2015, 271, 87–95 CrossRef CAS PubMed.
- K. Ullah, L. Zhu, Z.-D. Meng, S. Ye, Q. Sun and W.-C. Oh, Chem. Eng. J., 2013, 231, 76–83 CrossRef CAS PubMed.
- Z.-T. Hu, J. Liu, X. Yan, W.-D. Oh and T.-T. Lim, Chem. Eng. J., 2015, 262, 1022–1032 CrossRef CAS PubMed.
- B. S. Kwak, H. Lee and M. Kang, Chem. Eng. J., 2014, 255, 613–622 CrossRef CAS PubMed.
- L. Wang, J. Zhu, H. Yang, F. Wang, Y. Qin, T. Zhao and P. Zhang, J. Alloys. Compd., 2015, 634, 232–238 CrossRef CAS PubMed.
- M. Veerapandian, X. X. Zhu and S. Giasson, J. Mater. Chem. B, 2015, 3, 665–672 RSC.
- Y. Wu, W. Xu, Y. Wang, Y. Yuan and R. Yuan, Electrochim. Acta, 2013, 88, 135–140 CrossRef CAS PubMed.
- Y. Chen, X. Zhang, D. Zhang and Y. Ma, J. Alloys. Compd., 2012, 511, 251–256 CrossRef CAS PubMed.
- W. Wang, S. Guo, I. Lee, K. Ahmed, J. Zhong, Z. Favors, F. Zaera, M. Ozkan and C. S. Ozkan, Sci. Rep., 2014, 4, 4452 Search PubMed.
- C. Yu, W. Ji, Y. Wang, N. Bao and H. Gu, Nanotechnology, 2013, 24, 115502 CrossRef PubMed.
- C. Liu, J. Zhang, E. Yifeng, J. Yue, L. Chen and D. Li, Electron. J. Biotechnol., 2014, 17, 183–188 CrossRef CAS PubMed.
- E. Tully, S. P. Higson and R. O'Kennedy, Biosens. Bioelectron., 2008, 23, 906–912 CrossRef CAS PubMed.
- Centers for Disease Control and Prevention Online, http://www.cdc.gov/listeria/outbreaks/ice-cream-03-15/, (accessed August 2015).
- R. Wang, C. Ruan, D. Kanayeva, K. Lassiter and Y. Li, Nano Lett., 2008, 8, 2625–2631 CrossRef CAS PubMed.
- R. Radhakrishnan, M. Jahne, S. Rogers and I. I. Suni, Electroanalysis, 2013, 25, 2231–2237 CrossRef CAS PubMed.
- C. Cheng, Y. Peng, J. Bai, X. Zhang, Y. Liu, X. Fan, B. Ning and Z. Gao, Sens. Actuators, B, 2014, 190, 900–906 CrossRef CAS PubMed.
- M. Hirata, T. Gotou, S. Horiuchi, M. Fujiwara and M. Ohba, Carbon, 2004, 42, 2929–2937 CAS.
- M. Veerapandian, S. Sadhasivam, J. H. Choi and K. S. Yun, Chem. Eng. J., 2012, 209, 558–567 CrossRef CAS PubMed.
- M. Veerapandian, S. K. Lim, H. M. Nam, G. Kuppannan and K. S. Yun, Anal. Bioanal. Chem., 2010, 393, 867–876 CrossRef PubMed.
- P. Innocenzi, H. Kozuka and T. Yoko, J. Phys. Chem. B, 1997, 101, 2285–2291 CrossRef CAS.
- K. Mori, M. Kawashima, M. Che and H. Yamashita, Angew. Chem., Int. Ed., 2010, 49, 8598–8601 CrossRef CAS PubMed.
- S. Shukla and S. Saxena, Appl. Phys. Lett., 2011, 98, 073104 CrossRef PubMed.
- K. Krishnamoorthy, R. Mohan and S.-J. Kim, Appl. Phys. Lett., 2011, 98, 2441011 CrossRef PubMed.
- S. J. Jeon, S. Y. Kwak, D. B. Yim, J. M. Ju and J. H. Kim, J. Am. Chem. Soc., 2014, 136, 10842–10845 CrossRef CAS PubMed.
- P. Kannan, T. Maiyalagan, N. G. Sahoo and M. Opallo, J. Mater. Chem. B, 2013, 1, 4655–4666 RSC.
- J. Coates, in Encyclopedia of Analytical Chemistry, ed. R. A. Meyers, John Wiley & Sons, Chichester, 2000, pp. 10815–10837 Search PubMed.
- J. D. Liao, S.-P. Lin and Y. T. Wu, Biomacromolecules, 2005, 6, 392–399 CrossRef CAS PubMed.
- M. Veerapandian, M.-H. Lee, K. Krishnamoorthy and K. S. Yun, Carbon, 2012, 50, 4228–4238 CrossRef CAS PubMed.
- H. Friebolin, Basic One- and Two-Dimensional NMR Spectroscopy, Wiley-VCH, Weinheim, 5th edn, 2010 Search PubMed.
- R. S. Macomber, A Complete Introduction to Modern NMR Spectroscopy, Wiley, New York, 1998 Search PubMed.
- L. Malard, M. Pimenta, G. Dresselhaus and M. Dresselhaus, Phys. Rep., 2009, 473, 51–87 CrossRef CAS PubMed.
- F. Tuinstra and J. L. Koenig, J. Chem. Phys., 1970, 53, 1126–1130 CrossRef CAS PubMed.
- G. Jiang, Z. Lin, C. Chen, L. Zhu, Q. Chang, N. Wang, W. Wei and H. Tang, Carbon, 2011, 49, 2693–2701 CrossRef CAS PubMed.
- J. Shen, B. Yan, M. Shi, H. Ma, N. Li and M. Ye, J. Colloid Interface Sci., 2011, 356, 543–549 CrossRef CAS PubMed.
- M. Veerapandian, R. Subbiah, G. S. Lim, S. H. Park, K. S. Yun and M.-H. Lee, Langmuir, 2011, 27, 8934–8942 CrossRef CAS PubMed.
- Y. J. Choi and T.-J. M. Luo, Int. J. Electrochem., 2011, 404937 Search PubMed.
- L. Guo, J. Nie, B. Du, Z. Peng, B. Tesche and K. Kleinermanns, J. Colloid Interface Sci., 2008, 319, 175–181 CrossRef CAS PubMed.
- R. Pethig, Dielectric and Electronic Properties of Biological Materials, J. Wiley, New York, 1979 Search PubMed.
- L. Yang, Y. Li and G. F. Erf, Anal. Chem., 2004, 76, 1107–1113 CrossRef CAS PubMed.
- H. Sharma and R. Mutharasan, Biosens. Bioelectron., 2013, 45, 158–162 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra15329h |
| This journal is © The Royal Society of Chemistry 2015 |