
Formation and growth mechanisms of natural metastable twin boundary in crystalline β-octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine: a computational study†
Abstract
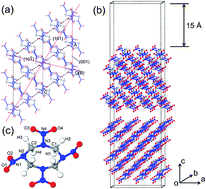
The twin boundary (TB), a typical planar defect, occurs naturally in molecular explosives and manipulates their sensitivities to external stimuli. We systemically studied the formation and growth mechanisms of the TBs in the β-octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine (β-HMX) crystal by using self-consistent charge density functional tight binding with dispersion corrections. The three TB species along the [010] zone axis based upon the experiments were considered. The (001)/[010] TB species are more favorable energetically than other TB species. The twinning in β-HMX most probably occurs on the (001) plane rather than on the (101) plane, which reproduces the naturally occurred twinned crystals in the experiments well. The TB-induced symmetry breaking alters not only the geometries but also on the electronic structures of the HMX molecules located at TB. The inner surface, outer surface, and intersection of the two surfaces are suggested to play vital roles in sensitizing the condensed phase β-HMX and to act as a trigger in initiating the chemical decomposition. The HMX molecule is most likely to be adsorbed in the concave site on (001) plane through either normal or twinning pathway in a competitive manner. After the grooves on (001) surface being filled, new grooves merge naturally for further adsorption.
 Please wait while we load your content...
Please wait while we load your content...

