
Poly(sodium 4-styrenesulfonate) modified graphene for reinforced biodegradable poly(ε-caprolactone) nanocomposites†
Abstract
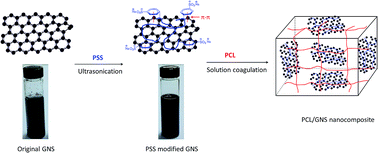
A homogeneous and stable water dispersion of graphene nanosheets (GNS) was prepared through a non-covalent functionalization technique by ultrasonic processing of GNS in the presence of poly(sodium 4-styrenesulfonate) (PSS) as the modifier. The dispersion was then used to compound with poly(ε-caprolactone) (PCL) through solution coagulation to fabricate PCL/GNS nanocomposites. Scanning and transmission electron microscope observations indicated that PSS modified GNS dispersed uniformly in and showed strong interfacial adhesion with the PCL matrix when the loading of GNS was not more than 0.5 wt%. While when the loading of GNS increased to 1.0 wt%, aggregation morphology of the GNS in the PCL matrix was detected. The crystallization temperature of PCL, as investigated using a differential scanning calorimeter, increased apparently by incorporation of PSS modified GNS. Investigation on isothermal crystallization kinetics of neat PCL and its composites indicated that the crystallization of PCL was accelerated considerably. Addition of only 0.05 wt% GNS caused a 5.8 times improvement in crystallization rate compared to neat PCL. The crystallization mechanism almost remained unchanged. The improvement in crystallization rate was ascribed to the enhanced nucleation density by incorporation of PSS modified GNS, as evidenced using a polarizing optical microscope (POM). Tensile tests showed that both the tensile strength and the Young's modulus of PCL were reinforced and increased gradually with increasing GNS loading within 0.5 wt%, meanwhile the elongation at break did not reduce but increased slightly. While when the loading of GNS increased to 1.0 wt%, the tensile strength and elongation at break reduced considerably due to the aggregation of GNS with increasing loading. Dynamic mechanical analysis indicated that the storage modulus of PCL was reinforced in the full temperature range by incorporation of PSS modified GNS.
 Please wait while we load your content...
Please wait while we load your content...

