
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polymers of intrinsic microporosity as high temperature templates for the formation of nanofibrous oxides†
H. Al Kutubiab,
L. Rassaeib,
W. Olthuisc,
G. W. Nelsond,
J. S. Foorde,
P. Holdwayf,
M. Cartag,
R. Malpass-Evansg,
N. B. McKeowng,
S. C. Tsangh,
R. Castainga,
T. R. Fordera,
M. D. Jonesa,
D. Hea and
F. Marken*a
aDepartment of Chemistry, University of Bath, Claverton Down, Bath BA2 7AY, UK. E-mail: f.marken@bath.ac.uk
bDepartment of Chemical Engineering, Delft University of Technology, Delft, The Netherlands
cBIOS/Lab-on-a-Chip group, University of Twente, PO Box 217, 7500 Enschede, The Netherlands
dImperial College London, Department of Materials, Royal School of Mines, Exhibition Road, London, SW7 2AZ, UK
eChemistry Research Laboratories, Oxford University, South Parks Road, Oxford OX1 3TA, UK
fDepartment of Materials, Begbroke Science Park, Sandy Lane, Yarnton, Oxford, OX5 1PF, UK
gSchool of Chemistry, University of Edinburgh, West Mains Road, Edinburgh, EH9 3JJ, UK
hInorganic Chemistry Laboratory, University of Oxford, South Parks Road, Oxford OX1 3QR, UK
First published on 24th August 2015
Abstract
The highly rigid molecular structure of Polymers of Intrinsic Microporosity (PIM) – associated with a high thermolysis threshold – combined with the possibility to fill intrinsic micropores allows the direct “one-step” templated conversion of metal nitrates into nano-structured metal oxides. This is demonstrated here with PIM-EA-TB and with PIM-1 for the conversion of Pr(NO3)3 to Pr6O11.
Nano-templating offers rapid access to novel nano-structured materials and interfaces1 in particular for technologies where high surface area inorganic architectures are desirable.2 Template hosts such as regular opaloid structures,3 surfactant-based nano-structures,4 or novel MOF structures5 have been proposed. Here, polymers of intrinsic microporosity (PIM)6 are introduced as “high temperature templates” for “one-step” metal oxide nano-structure growth as demonstrated for the case of Pr6O11.
Polymers of intrinsic microporosity represent a novel group of polymers with a rigid backbone (see structures in Fig. 1) that prevents them from collapsing into a close-packed conformation even when heated up. Space is created within the polymer, allowing for permanent microporosity and leading to a surface area as high as 900 m2 g−1 for PIM-1 (ref. 7) and 1027 m2 g−1 for PIM-EA-TB.8 PIM materials are readily casted from solution into films and have been investigated for applications in gas separation membranes, catalysis, and gas storage.9 From thermogravimetric data (TGA, see Fig. 1) it is clear that these rigid polymer structures also show considerable high temperature stability (after some initial weight loss due to water desorption below 100 °C, decomposition onset occurs for PIM-EA-TB at 310 °C and for PIM-1 at 480 °C, both with charring). Therefore, in this study we contrast the ability of PIM-EA-TB and PIM-1 to function as template hosts for high temperature metal oxide nanostructure synthesis. A suitable model nano-structured metal oxide with promise for application in sensors10 and in catalysis11 is Pr6O11.
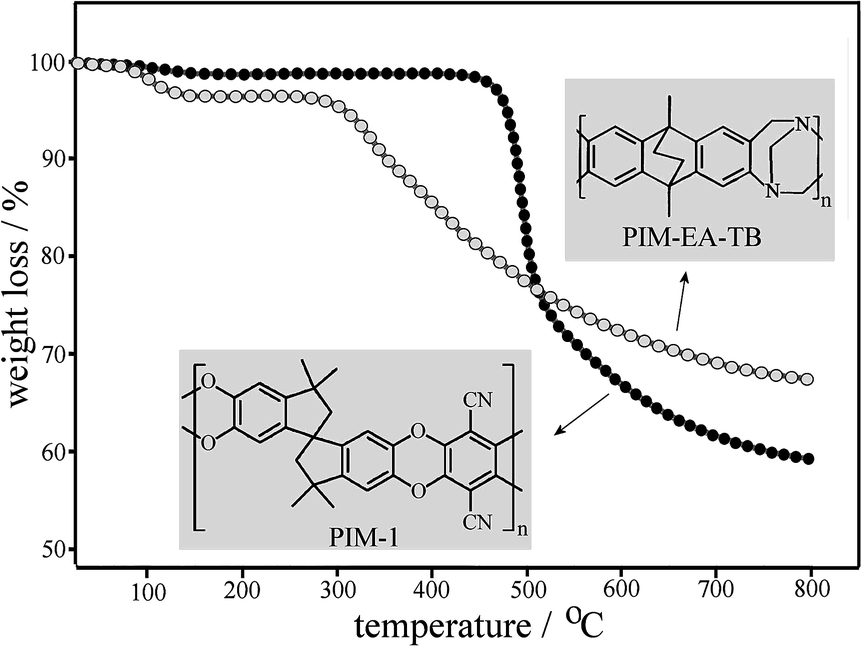 | ||
| Fig. 1 Thermogravimetric data and molecular structures for PIM-EA-TB and PIM-1. | ||
The synthesis of praseodymium oxides has been carried out previously by chemical vapour deposition,12 calcination of praseodymium hydroxide (Pr(OH)3),13,14 electro-deposition,15 or by thermal transformation of a praseodymium-containing precursor compound.16 The products obtained through thermal oxidation depend on both the precursor as well as oxidation conditions such as temperature and oxygen partial pressure. The oxygen deficient Pr6O11 phase can be formed as dominant phase at temperatures higher than 465 °C.17 Bäumer and coworkers11b investigated the formation of nanostructured praseodymium oxide via thermal decomposition of praseodymium nitrate with and without carbon-based templates. Here, we report the formation of praseodymium oxide structures not in bulk, but directly at the surface of tin-doped indium oxide (ITO) electrodes.
When the PIM host solution (1 mg mL−1 in chloroform) and Pr(NO3)3 solution (1 mg mL−1 in DMF) are mixed in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 weight ratio and deposited onto ITO, calcination at 500 °C in air affords a thin film of oxide materials on ITO (see Experimental, Fig. 2).
:1 weight ratio and deposited onto ITO, calcination at 500 °C in air affords a thin film of oxide materials on ITO (see Experimental, Fig. 2).
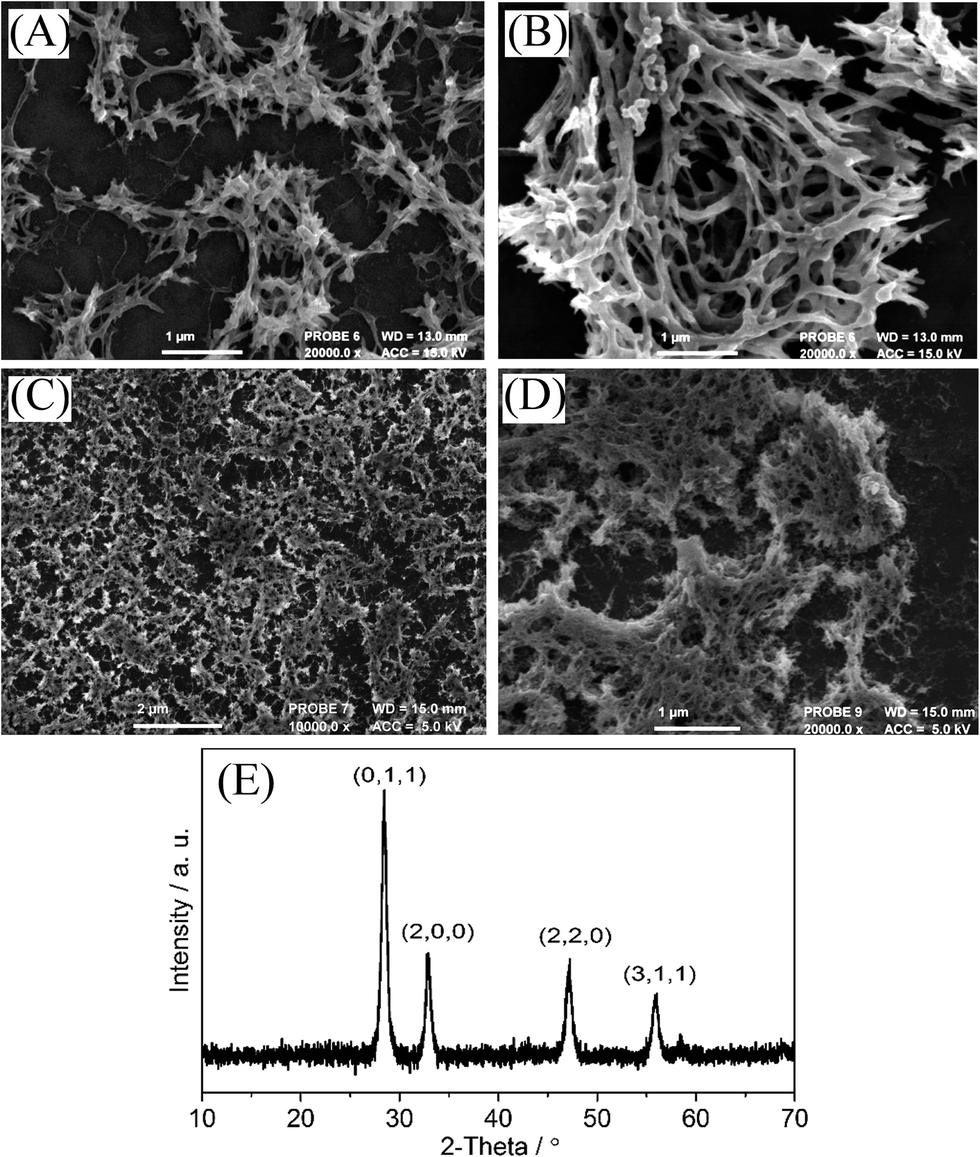 | ||
| Fig. 2 Electron micrographs of (A) a 10-layer PIM-EA-TB – Pr(NO3)3 (1:1) film after calcination, (B) a 20-layer PIM-EA-TB – Pr(NO3)3 (1:1) film after calcination, (C) a 10-layer PIM-1 – Pr(NO3)3 (1:1) film after calcination and (D) a higher resolution image of the same sample. (E) XRD analysis. | ||
The presence of praseodymium oxide is confirmed by EDX (see Fig. S1D†) and by XRD (Fig. 2E, with characteristic lines15,11b). Electron micrographs show fibrous deposits of Pr6O11 on the ITO substrate (Fig. 2). With PIM-EA-TB as host template “leaf-like” nano-structures are seen. Doubling the amount of precursor deposit resulted in slightly courser structures, which must reflect the pore geometry of the precursor at the point when solidification of the oxide precursor occurs. Changing the ratio of PIM-EA-TB to Pr(NO3)3 resulted in similar structures (see Fig. S1A–C†). When investigating the PIM-1 template (see Fig. 2C and D) it became apparent that a much finer nano-structure with filaments down to 20 nm or less are formed. BET-based pore size data for PIM-EA-TB polymer (12–40 Å (ref. 18)) and for PIM-1 polymer (5 to 15 Å (ref. 19)) suggest that in both the parent polymers only comparably smaller pores are present. The feature size in the Pr6O11 deposits appear considerably bigger for PIM-EA-TB but more similar to the original pore size for PIM-1. Therefore the feature size could be linked to the behaviour of the polymer template at elevated temperature. TGA data in Fig. 1 clearly show the higher thermal stability of PIM-1, which is likely to result in a finer oxide nano-structure that more closely reflects the original PIM-1 template pores.
In order to demonstrate the absence of polymer remnants, further surface analysis has been performed with XPS (Fig. 3). Apart from the underlying ITO surface elements clear evidence for Pr, C, and O is observed in the survey scan. Carbon signals are very low and assigned to adventitious surface-adsorbed molecules (or possibly remnants of the template). Oxygen signals are assigned predominantly to Pr6O11, but with some other species present at the surface. Wolffram et al.20 have studied thin PrxOy films made from Pr6O11 targets and their work is the primary basis for fitting the O1s spectra here. Four peaks were required to curve fit the O1s spectra. The two main component at ≈528.5 eV and ≈531 eV most likely belong to Pr2O3 and Pr6O11, respectively. Lutkehoff et al.21 indicated that the signal at ≈532 eV can be ascribed to Pr-based hydroxides, such as Pr(OH)3. These would be expected from the breakdown of Pr6O11 in the presence of surface water (eventually leading to PrO2 formation).20 The feature at ≈529.5 eV could be either indicative of the presence of PrO2 (ref. 22) or be related to surface adsorbates in the form of Pr-O-R;20 both species have O1s signal known to overlap with the Pr6O11 O1s signal. As seen in Table 1, the Pr6O11 and Pr-hydroxide content at surfaces seem independent of the method of preparation. However, use of PIM-EA-TB favours the additional formation of Pr2O3 (Pr3+), whilst use of PIM-1 favours the formation of PrO2 (Pr4+) and surface adsorbates. The chemical reasons for this difference are currently not fully understood.
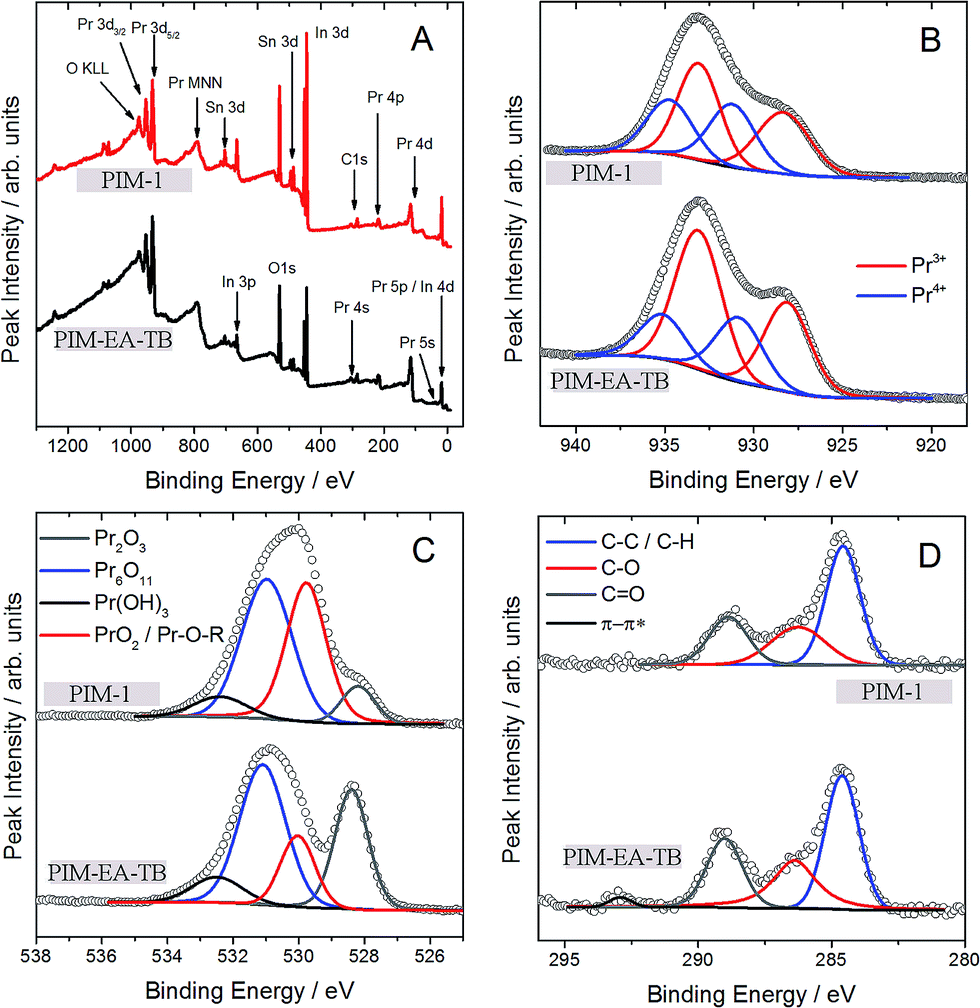 | ||
| Fig. 3 XPS survey spectra (A) of PIM-EA-TB and PIM-1 and core level data for (B) Pr3d5/2 (with red and blue curve fits for Pr3+ and Pr4+ species, respectively), (C) O1s, (D) C1s. | ||
| Species | % O1s composition | |
|---|---|---|
| PIM-EA-TB | PIM-1 | |
| Pr2O3 | 29.2 | 8.2 |
| PrO2/adsorbates | 16.7 | 39.2 |
| Pr6O11 | 45.3 | 45.8 |
| Hydroxides | 8.8 | 6.9 |
Wolffram et al.20 note that unambiguous fitting of the Pr3d core levels is difficult and remains controversial. Again using the above reference as a model, four chemical environments were curve fitted. The line pair at higher binding energy (≈931 eV and ≈935 eV) are ascribed to Pr4+ species (e.g. Pr6O11 and PrO2). Assuming that they reflect chemical environments at the surface and are not satellites.20 The line pair at lower binding energy (≈928 eV and ≈933 eV) originate from Pr3+ (e.g. Pr2O3 and Pr(OH)3). The ratio between Pr3+:Pr4+ species is approximately 2:1 and 4:3 for PIM-EA-TB and PIM-1, respectively. Notwithstanding the different chemical states, the surface O/Pr ratio is 4.6 and 2.6 for PIM-EA-TB and PIM-1, respectively. These ratios are quite high – the stoichiometric and expected O/Pr ratio for Pr2O3 (Pr3+) and Pr6O11 (Pr4+) is 1.5 and 1.8, respectively. This may indicate that the O1s signal is influenced by other sources of surface oxygen, other than the Pr-oxides (e.g. hydroxides, water, the underlying substrate, etc.). One expects an O/Pr ratio <2:1 for the Pr-based oxides.20 It is clear from Table 2 that PIM-EA-TB has a higher Pr3+ component than PIM-1, and vice versa in the case of the Pr4+ species. In future, bulk elemental analytical methods have to be employed to further investigate bulk phase purity and possible impurities from the thermolysis process in the resulting products as a function of thermolysis time and temperature.
| Pr3d5/2 binding energy (eV) | Pr3d composition (%) | |||||
|---|---|---|---|---|---|---|
| 928 | 931 | 933 | 935 | Pr3+ | Pr4+ | |
| PIM-EA-TB | 27.0 | 19.2 | 39.8 | 14.0 | 66.8 | 33.2 |
| PIM-1 | 25.9 | 23.1 | 31.5 | 19.5 | 57.4 | 42.6 |
Electrochemical testing of Pr6O11 nano-structures was performed in aqueous 0.1 M KNO3 (Fig. 4). Nyquist plots (Fig. 4A) and Bode plots (not shown) suggest a high frequency switch from resistive to capacitive behaviour associated with the ITO substrate time constant (for bare ITO 110 Ω × 15 μF = 1.65 ms, Fig. 4D). With Pr6O11 films deposited an additional resistive component, Ret, is observed associated with charging of the nano-structured deposit. The impedance for this charging decreases from a calcined 10-layer PIM-EA-TB – Pr(NO3)3 deposit to calcined 20-layer PIM-EA-TB – Pr(NO3)3 and again to calcined 10-layer PIM-1 – Pr(NO3)3. This result indicates an increase in oxide surface area in this sequence.
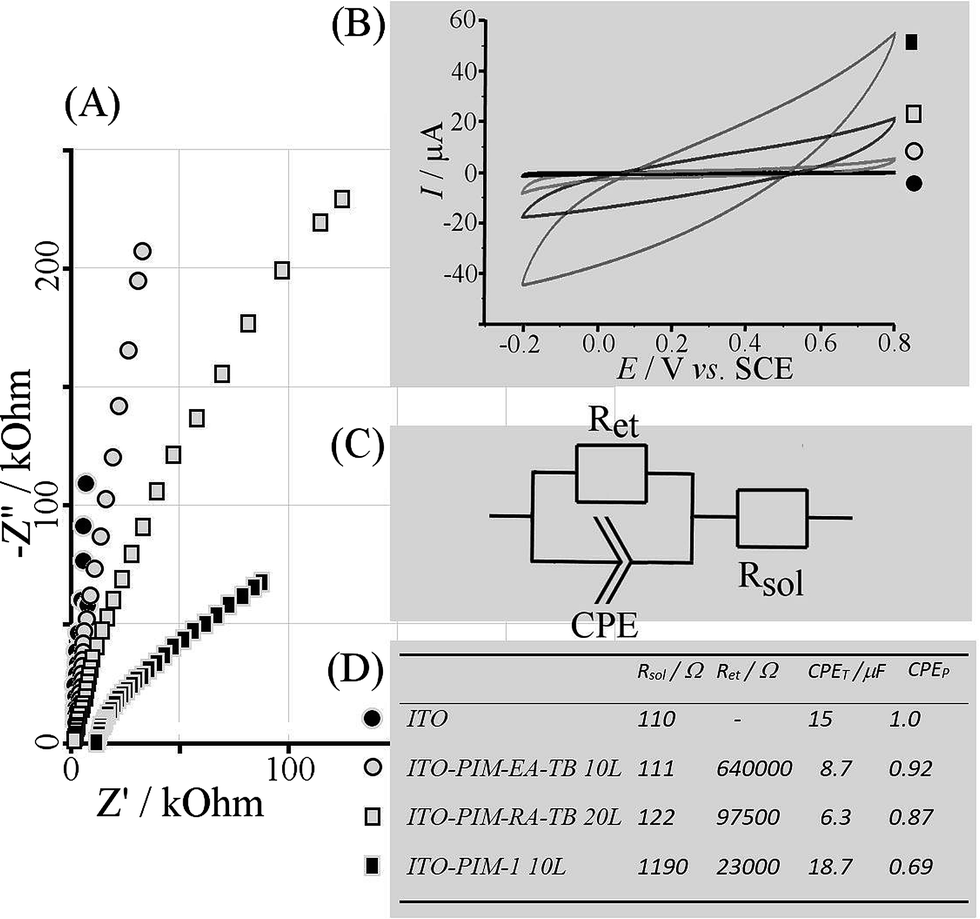 | ||
| Fig. 4 (A) Nyquist plots (1 Hz to 128 kHz) with impedance data for bare ITO, PIM-EA-TB 10-layer, PIM-EA-TB 20-layer, and PIM-1 10-layer deposits biased at 0.3 V vs. SCE in 0.1 M KNO3. (B) Cyclic voltammograms (scan rate 200 mVs−1) under the same conditions. (C) Equivalent circuit and (D) summary of data. | ||
Complementary cyclic voltammetry data (Fig. 4B) also demonstrate the decrease in impedance as an increase in charging current. Full charging and therefore full capacitive characteristics would require more time (or a higher conductivity of the oxide). The electrochemical properties are consistent with those reported previously for Pr6O11 with potential applications in charge storage and sensing. However, the methodology for oxide nano-structure formation in PIM templates will be applicable for a much wider range of oxides and mixed oxides.
Experimental
Chemical reagents
Praseodymium nitrate hexa-hydrate, N,N-dimethylformamide (DMF) and chloroform were obtained from Sigma-Aldrich and used without further purification. Polymers with intrinsic microporosity PIM-EA-TB8 and PIM-1 (ref. 23) were prepared following literature procedures. Tin-doped indium oxide glass plates (ITO) with a resistivity of 15 Ω per square were obtained from Image Optics Components Ltd (Basildon, UK). A KCl-saturated calomel (SCE) reference electrode was obtained from radiometer.Instrumentation
The morphology of the samples was analysed using a JEOL FESEM6301F field emission scanning electron microscopy (FE-SEM). XPS experiments were conducted using a Thermo K Alpha (Thermo Scientific) spectrometer (operating at ≈10−8–10−9 Torr) with a 180° double focusing hemispherical analyser running in constant analyser energy (CAE) mode and a 128-channel detector. A mono-chromated Al Kα radiation source (1486.7 eV) was used. Peak fitting was conducted using XPS Peak Fit (v. 4.1) software using Shirley background subtraction. Peaks were referenced to the adventitious carbon C1s peak (284.6 eV) and peak areas were normalized to the photoelectron cross-section of the F1s photoelectron signal using atomic sensitivity factors.24 An Elite Thermal Systems Ltd tube furnace was used to remove the possible organic contamination on the ITO electrodes and for calcination of metal oxides. Electrochemical testing was performed using an Ecochemie Autolab PGSTAT12 potentiostat system. TGA data were collected on a Setaram Setsys Evolution TGA instrument. The samples were heated under Ar from 20 °C until 800 °C at 10 K per minute.Procedure for nano-Pr6O11 film deposition
Tin-doped indium oxide (ITO) coated glass slides were cut into 1 cm × 3 cm strips and cleaned by rinsing with water and ethanol, followed by calcination at 500 °C for one hour. A solution of 1 mg mL−1 PIM in chloroform was mixed with a solution of 1 mg mL−1 Pr(NO3)3·6H2O in DMF in the desired ratio. From the resulting mixture, 25 μL was deposited onto a clean ITO plate covering approximately 1 cm2 and dried in an oven at 100 °C for 15 minutes. This deposition process was repeated for a desired number of layers and finally followed by calcination in a tube furnace at 500 °C for 1 hour.Conclusions
Praseodymium oxide nano-structures have been formed in a convenient and novel “one-step” process using a high temperature template based on polymers of intrinsic microporosity. The resulting structures differ from those obtained through simple calcination and show a leaf-like or nano-fibrous structures. Finer structures are formed with the more thermally stable PIM-1 template. The results indicate that this methodology could be used beneficially for the rapid formation of a wider range of nano-structured metal oxide as well as mixed metal oxides with future applications in electronic, sensor, or solar cell components.Notes and references
- I. W. Hamley, Nanotechnology, 2003, 14, R39 CrossRef CAS.
- P. V. Braun, P. Osenar, V. Tohver, S. B. Kennedy and S. I. Stupp, J. Am. Chem. Soc., 1999, 121, 7302 CrossRef CAS.
- A. Stein, Microporous Mesoporous Mater., 2001, 44, 227 CrossRef.
- G. J. D. Soler-illia, C. Sanchez, B. Lebeau and J. Patarin, Chem. Rev., 2002, 102, 4093 CrossRef PubMed.
- X. Xu, R. Cao, S. Jeong and J. Cho, Nano Lett., 2012, 12, 4988 CrossRef CAS PubMed.
- N. B. McKeown and P. M. Budd, Chem. Soc. Rev., 2006, 35, 675 RSC.
- P. M. Budd, B. S. Ghanem, S. Makhseed, N. B. McKeown, K. J. Msayib and C. E. Tattershall, Chem. Commun., 2004, 230 RSC.
- M. Carta, R. Malpass-Evans, M. Croad, Y. Rogan, J. C. Jansen, P. Bernardo, F. Bazzarelli and N. B. McKeown, Science, 2013, 339, 303 CrossRef CAS PubMed.
- N. B. McKeown and P. M. Budd, Macromolecules, 2010, 43, 5163 CrossRef CAS.
- (a) X. S. Liang, Y. H. He, F. M. Liu, B. Wang, T. G. Zhong, B. Quan and G. Lu, Sens. Actuators, B, 2007, 125, 544 CrossRef CAS PubMed; (b) S. Shrestha, C. M. Y. Yeung, C. E. Mills, J. Lewington and S. C. Tsang, Angew. Chem., Int. Ed., 2007, 46, 3855 CrossRef CAS PubMed.
- (a) E. S. Putna, J. M. Vohs, R. J. Gorte and G. W. Graham, Catal. Lett., 1998, 54, 17 CrossRef CAS; (b) Y. Borchert, P. Sonstrom, M. Wilhelm, H. Borchert and M. Bäumer, J. Phys. Chem. C, 2008, 112, 3054 CrossRef CAS.
- H. C. Aspinall, J. Gaskell, P. A. Williams, A. C. Jones, P. R. Chalker, P. A. Marshall, J. F. Bickley, L. M. Smith and G. W. Critchlow, Chem. Vap. Deposition, 2003, 9, 235 CrossRef CAS PubMed.
- X. W. Ouyang, S. S. Yuan, Q. B. Qiu, W. W. Zeng, G. A. Hope and H. H. Li, Inorg. Chem. Commun., 2014, 46, 21 CrossRef CAS PubMed.
- S. Shrestha, C. M. Y. Yeung, F. Marken, C. E. Mills and S. C. Tsang, Sens. Actuators, B, 2007, 123, 400 CrossRef CAS PubMed.
- S. Shrestha, F. Marken, J. Elliott, C. M. Y. Yeung, C. E. Mills and S. C. Tsang, J. Electrochem. Soc., 2006, 153, C517 CrossRef CAS PubMed.
- S. Zinatloo-Ajabshir and M. Salavati-Niasari, New J. Chem., 2015, 39, 3948 RSC.
- G. A. M. Hussein, B. A. A. Balboul, M. A. A. Warith and A. G. M. Othman, Thermochim. Acta, 2001, 369, 59 CrossRef CAS.
- E. Madrid, Y. Y. Rong, M. Carta, N. B. McKeown, R. Malpass-Evans, G. A. Attard, T. J. Clarke, S. H. Taylor, Y. T. Long and F. Marken, Angew. Chem., Int. Ed., 2014, 53, 10751 CrossRef CAS PubMed.
- M. Heuchel, D. Fritsch, P. M. Budd, N. B. McKeown and D. Hofmann, J. Membr. Sci., 2008, 318, 84 CrossRef CAS PubMed.
- D. Wolffram, M. Ratzke, M. Kappa, M. J. Montenegro, M. Döbeli, Th. Lippert and J. Reif, J. Mater. Sci. Eng. B, 2004, 109, 24 CrossRef PubMed.
- S. Lutkehoff, M. Neumann and A. Slebarski, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 52, 13808 CrossRef.
- M. Sawangphruk and J. S. Foord, Diamond Relat. Mater., 2010, 19, 885 CrossRef CAS PubMed.
- (a) P. M. Budd, K. J. Msayib, C. E. Tattershall, B. S. Ghanem, K. J. Reynolds, N. B. McKeown and D. Fritsch, J. Membr. Sci., 2005, 251, 263 CrossRef CAS PubMed; (b) P. M. Budd, E. S. Elabas, B. S. Ghanem, S. Makhseed, N. B. McKeown, K. J. Msayib, C. E. Tattershall and D. Wang, Adv. Mater., 2004, 16, 456 CrossRef CAS PubMed.
- C. D. Wagner, L. E. Davis, M. V. Zeller, J. A. Taylor, R. M. Raymond and L. H. Gale, Surf. Interface Anal., 1981, 3, 211 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional scanning electron microscopy data and EDX data. See DOI: 10.1039/c5ra15131g |
| This journal is © The Royal Society of Chemistry 2015 |