
Synthesis, characterization and in vitro cytotoxicity of gold(iii) dialkyl/diaryldithiocarbamato complexes†
Abstract
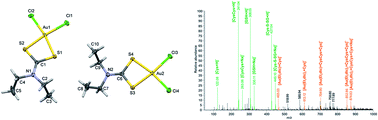
A series of six dialkyl/diaryldithiocarbamato (dtc) gold(III) complexes 1–6 of the composition [Au(Me2dtc)2]Cl (1), [Au(Me2dtc)Cl2] (2), [Au(Et2dtc)2]Cl (3), [Au(Et2dtc)Cl2] (4), [Au(Bn2dtc)2]Cl (5), [Au(Bn2dtc)Cl2] (6) (dtc = Dithiocarbamate, Me = Methyl, Et = Ethyl and Bn = Benzyl) was synthesised and characterized by elemental analysis, infrared and NMR spectroscopy, and single crystal X-ray analysis in the case of complexes 4 and 5. The anticancer potential of the complexes was evaluated against MCF7, A2780 and A2780R human cancer cell lines as well as against a healthy MRC5 cell line. The most promising data were obtained for complex 4. It showed similar cytotoxicity as cisplatin against A2780 cells (EC50 ≈ 9.5 μM vs. EC50 ≈ 10 μM), and significantly higher cytotoxicity as compared to cisplatin on A2780R and MCF7 cell lines, with EC50 ≈ 2.8 μM vs. EC50 ≈ 21 μM, and EC50 ≈ 2.2 μM vs. EC50 > 50 μM, respectively. The interactions of the representative complexes 3 and 4 with a mixture of physiological levels of L-cysteine (Cys) and reduced L-glutathione (GSH) were also studied.
 Please wait while we load your content...
Please wait while we load your content...

