
Solid state NMR characterization of zeolite beta based drug formulations containing Ag and sulfadiazine†
Abstract
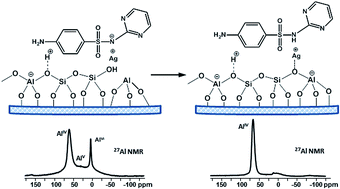
In this study we present a detailed characterization of zeolite beta-based drug formulations with antibacterial properties, containing Ag and sulfadiazine (SD). Solid state NMR spectroscopy (1H, 13C, 29Si, 27Al and 1H–29Si CP-HETCOR experiments) was applied to address some important questions concerning the changes in zeolite structure as a result of the simultaneous presence of both drugs SD and silver sulfadiazine (AgSD). A mechanism for transformation of octahedral defect framework Al sites and encapsulation of the extraframework Al (EFAl) present in the parent beta material into framework tetrahedral species as a result of the drug loading procedure is proposed. The analysis of 1H spectra suggested that an ion exchange process between the zeolite protons and Ag ions occurred upon the solid-state procedure of silver introduction by AgNO3 or AgSD. Suggestions about the location of the drug molecules inside the pores and/or on the crystallite surface are made and the nature of the drug–carrier interactions is discussed. Comparison of 1H spin-lattice (T1) and spin–spin (T2) relaxation times of pure drugs and drug loaded formulations indicate amorphization of the drug incorporated into the zeolite pores.
 Please wait while we load your content...
Please wait while we load your content...

