
Targeting a chemorefractory COLO205 (BRAF V600E) cell line using substituted benzo[α]phenoxazines†
Sanjima
Pal
a,
V. Badireenath
Konkimalla
*a,
Laxmi
Kathawate
b,
Soniya S.
Rao
b,
Shridhar P.
Gejji
b,
Vedavati G.
Puranik
c,
Thomas
Weyhermüller
d and
Sunita
Salunke-Gawali
*b
aSchool of Biological Sciences, National Institute of Science Education and Research (NISER), Bhubaneswar 751005, Orissa, India. E-mail: badireenath@niser.ac.in
bDepartment of Chemistry, Savitribai Phule Pune University, Pune 411007, India. E-mail: sunitas@chem.unipune.ac.in; Fax: +91 2025693981; Tel: +91 2025601397 ext. 531
cCenter for Material Characterization, National Chemical Laboratory, Pune 411008, India
dMPI für Chemische Energiekonversion, Stiftstr. 34-36, 45470 Mülheim an der Ruhr, Germany
First published on 14th September 2015
Abstract
Mutational activations of the oncogene BRAF (especially BRAF V600E) result in a poor prognosis for colon cancer patients and are associated with chemoresistance rendering them refractory to treatment. The development of novel bioactive compounds with specific targeting abilities under such conditions is an urgent need in drug discovery. In this report we synthesize and characterize three fluorescent benzo[α]phenoxazine compounds (10R-benzo[α]phenoxazine-5-one, 1B; R = Cl, 2B; R = CH3, 3B; R = H) and their anticancer activities are evaluated in a COLO205 cell line. All three compounds with a log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P value around 2 were cell permeable. However, 2B and 3B showed specific cytotoxicity in a malignant COLO205 cell line with a BRAF mutation (V600E) in comparison to a non-malignant wild-type BRAF HEK293T cell line. From further cell-based assays (cell cycle analysis, DNA fragmentation and caspase activation), we conclude that 2B and 3B treatment-induced selective cell death by inducing cell cycle arrest at the G0/G1 phase and caspase-mediated apoptosis (activation of the intrinsic and extrinsic pathways) are present only in BRAF V600E COLO205 cells. Further studies in the drug discovery pipeline might help develop these benzo[α]phenoxazines as promising chemotherapeutics for such refractory mutated cancers.
P value around 2 were cell permeable. However, 2B and 3B showed specific cytotoxicity in a malignant COLO205 cell line with a BRAF mutation (V600E) in comparison to a non-malignant wild-type BRAF HEK293T cell line. From further cell-based assays (cell cycle analysis, DNA fragmentation and caspase activation), we conclude that 2B and 3B treatment-induced selective cell death by inducing cell cycle arrest at the G0/G1 phase and caspase-mediated apoptosis (activation of the intrinsic and extrinsic pathways) are present only in BRAF V600E COLO205 cells. Further studies in the drug discovery pipeline might help develop these benzo[α]phenoxazines as promising chemotherapeutics for such refractory mutated cancers.
Introduction
RAS and RAF kinases are integral parts of the RAS–RAF–MAP2K (MEK)–MAPK signalling pathway that responds to several growth factors and cytokines.1–3 There are several reports available indicating the importance of aberrantly functional KRAS and BRAF protein kinases in tumor maintenance.2,4–6 Despite having a common activator (Ras) and substrate (MEK), the BRAF isoform has the highest activity among its three isoforms (A-,B- and C-RAF).7–10 A specific point mutation BRAF V600E (c.1799T->A) within the kinase activation domain of the BRAF protein is the most common BRAF mutation that constitutively activates substrate MEK eventually leading to development of several types of malignant and drug resistant cancers.11–14Epidemiological studies have demonstrated that almost 8% of all solid tumors including 50% of melanomas, 30–70% of papillary thyroid carcinomas and 5–8% of colorectal adenocarcinomas are associated with this particular V600E mutation.15 A dramatic response rate was observed when malignant melanoma cells with BRAF V600E mutation were treated with vemurafenib (a BRAF V600E selective inhibitor), however, a low clinical response was reported in colon cancer patients bearing the same mutation.16–18 Most colon cancer cells harboring such a point mutation are highly metastatic and non-responsive to established treatment regimes e.g., an anti-EGFR mAb (cetuximab, panitumumab) and/or chemotherapeutic inhibitors (vemurafenib, sorafenib, or MEK inhibitors).19–22 Colon cancer cells acquire such characteristics by adopting different mechanisms e.g. amplification or altered splicing of BRAF genes, and an altered status of EGFR.13,23–25 With no suitable biomarkers available to date, a proper prognosis for BRAF V600E mutation in cancers is still elusive for treatment of colorectal cancer with standard chemotherapeutic agents or anti-EGFR monoclonal antibodies.20,26–29 Therefore understanding the importance and prevalence of the V600E BRAF mutation in chemoresistant cancers, there is a need to develop novel organic compounds that can cause cell death in a V600E BRAF mutation condition selectively.
In this context, the chemical structure of benzo[α]phenoxazine (BPZ) and its derivatives exhibit several interesting features for development as a suitable targeting molecule. Molecular docking studies with a G-tetrad showed that the delocalized π-electrons from the planar BPZ core-group promoted stable π-stacking with the G-tetrad of DNA.30 Biophysical studies using SPR further confirmed its selective affinity for DNA secondary structures, where BPZ binding to a G-quadruplex sequence in a c-KIT promoter and reducing c-KIT expression in a human gastric carcinoma cell line was reported indicating its potential as an anti-tumor agent.31 In a previous study, we reported the inhibitory activity of some BPZ derivatives for topoisomerases (DNA-binding enzymes).32 Apart from DNA binding, some derivatives of BPZ induced photocytotoxicity in murine sarcoma cells by sensing the pH in the microenvironment and changing the redox status within the cell.33 Furthermore, the inherent fluorescent nature of these compounds and their ability to permeate cells has shown them to selectively localize in different cell organelles (mitochondria and golgi) enabling cell imaging studies.34 This way BPZ derivatives offer a great advantage in cancer chemotherapy as theranostics where solid cancers can be specifically tracked and treated simultaneously.
In the current study, a BRAF mutated colon cancer cell line, COLO205, that demonstrates all the above mentioned treatment resistance characteristics was considered as a suitable model for screening new bioactive chemical entities under such mutation conditions.35,36 Here, three intrinsically fluorescent benzo[α]phenoxazines (10R-benzo[α]phenoxazine-5-one, 1B; R = Cl, 2B; R = CH3, 3B; R = H, Scheme 1) were synthesized and characterized. Their anti-proliferative activities were assessed on COLO205. These compounds being intrinsically fluorescent, enabled us to conduct further validation studies of their cellular uptake, distribution and dose-dependent activity effectively using flow cytometry and fluorescence microscopy.
 | ||
| Scheme 1 Molecular structures of 1B, 2B and 3B. | ||
Results and discussion
Synthesis
Benzo[α]phenoxazine derivatives 1B, 2B and 3B were synthesized by Michael addition of the phenolic hydroxyl groups of 4R-2-aminophenol (R = Cl; 1B, CH3; 2B and H; 3B) to 2-hydroxy-1,4-naphthoquinone.371B, 2B and 3B are the minor products of the reaction between 2-hydroxy-1,4-naphthoquinone and aminophenol derivatives. The reaction mechanism is presented in Scheme 2. The 1,4-Michael addition–elimination of the hydroxyl group of the aminophenol to the C(2) carbon of 2-hydroxy-1,4-naphthoquinone results in species (iv), and further 1,2-addition of the amino group of aminophenol to the carbonyl carbon C(1) generates an aminol as unstable intermediate (vi). Further dehydration of (vi) results in the final products 1B to 3B. | ||
| Scheme 2 Reaction mechanism involved in the synthesis of 1B to 3B. | ||
The molecular mass and purity of the compounds were determined using HR-MS (Fig. S1, S5 and S9 in the ESI†). The peaks at frequencies of 1647, 1639 and 1635 cm−1 in the FT-IR spectra were assigned to the νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibration in 1B, 2B and 3B respectively and the νCN frequency was assigned at 1573, 1587 and 1593 cm−1 respectively for 1B, 2B and 3B. A para-naphthoquinone (p-NQ) vibration (∼1280 cm−1) was present in all compounds, however a peak due to νC(6A)–O(7) was observed at ∼1240, 1226 and 1234 cm−1 for 1B, 2B and 3B respectively. Chemical shifts were assigned using 2D gHSQCAD experiments (Fig. S4, S8 and S12 in the ESI†). Unlike the quinone compounds, the benzo[α]phenoxazine derivatives 1B, 2B and 3B were redox active (Fig. S13 in the ESI†). The cyclic voltammograms of all the compounds showed one electron reversible redox couple at E1/2 = 0.297 V, −0.379 V and −0.442 V for 1B, 2B and 3B respectively in DMSO solution and the calculated logP (using chem sketch program) was determined to be 2.87, 2.79 and 2.33 respectively.
O vibration in 1B, 2B and 3B respectively and the νCN frequency was assigned at 1573, 1587 and 1593 cm−1 respectively for 1B, 2B and 3B. A para-naphthoquinone (p-NQ) vibration (∼1280 cm−1) was present in all compounds, however a peak due to νC(6A)–O(7) was observed at ∼1240, 1226 and 1234 cm−1 for 1B, 2B and 3B respectively. Chemical shifts were assigned using 2D gHSQCAD experiments (Fig. S4, S8 and S12 in the ESI†). Unlike the quinone compounds, the benzo[α]phenoxazine derivatives 1B, 2B and 3B were redox active (Fig. S13 in the ESI†). The cyclic voltammograms of all the compounds showed one electron reversible redox couple at E1/2 = 0.297 V, −0.379 V and −0.442 V for 1B, 2B and 3B respectively in DMSO solution and the calculated logP (using chem sketch program) was determined to be 2.87, 2.79 and 2.33 respectively.
Single crystal X-ray diffraction studies of 2B and 3B
Compounds 2B and 3B crystallize in monoclinic space group P21/n. Fig. 1 shows the ORTEP plots and the crystallography parameters are presented in Table 1. The bond distance of C(5)O(5) in both the compounds is ∼1.23 Å which typically indicates the oxidized form of quinonoid carbonyl.32,37–39 After forming planar polycycles, both the compounds retain their quinonoid distortion in ring B (Scheme 1) which was adjudged using the bond distances of CO and CN.
 | ||
| Fig. 1 ORTEP plots of 2B and 3B, the ellipsoid was drawn with 50% probability. | ||
| Identification code | 2B | 3B |
| Empirical formula | C17H11NO2 | C16H9NO2 |
| Formula weight | 261.27 | 247.24 |
| Temperature | 100(2) K | 293(2) K |
| Wavelength | 0.71073 Å | 0.71073 Å |
| Crystal system | Monoclinic | Monoclinic |
| Space group | P21/n | P21/n |
| Unit cell dimensions | a = 13.288(2) Å, b = 4.6447(6) Å, β = 101.696(2)°, c = 19.479(3) Å | a = 3.91910(9) Å, b = 23.3060(5) Å, β = 94.315(1)°, c = 12.3580(3) Å |
| Volume | 1177.3(3) Å3 | 1125.56(4) Å3 |
| Z | 4 | 4 |
| Density (calculated) | 1.474 Mg m−3 | 1.459 Mg m−3 |
| Absorption coefficient | 0.098 mm−1 | 0.097 mm−1 |
| F(000) | 544 | 512 |
| Crystal size | 0.834 × 0.072 × 0.061 mm3 | 0.38 × 0.09 × 0.04 mm3 |
| Theta range for data collection | 1.706 to 30.889° | 1.75 to 23.97° |
| Index ranges | −19 ≤ h ≤ 19, −6 ≤ k ≤ 6, −28 ≤ l ≤ 28 | −4 ≤ h ≤ 4, −26 ≤ k ≤ 25, −13 ≤ l ≤ 14, −7 ≤ h ≤ 8, — |
| Reflections collected | 31939 |
7783 |
| Independent reflections | 3718 [R(int) = 0.0516] | 1766 [R(int) = 0.0473] |
| Completeness to theta = 25.242° | 99.9% | 99.6% |
| Absorption correction | Gaussian | Semi-empirical from equivalents |
| Max. and min. transmission | 0.99490 and 0.95827 | 0.9961 and 0.9636 |
| Refinement method | Full-matrix least-squares on F2 | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 3718/0/182 | 1766/0/209 |
| Goodness-of-fit on F2 | 1.076 | 1.041 |
| Final R indices [I > 2sigma(I)] | R 1 = 0.0408, wR2 = 0.1144 | R 1 = 0.0350, wR2 = 0.0901 |
| R indices (all data) | R 1 = 0.0514, wR2 = 0.1251 | R 1 = 0.0508, wR2 = 0.1019 |
| Extinction coefficient | n/a | 0.015(3) |
| Largest diff. peak and hole | 0.497 and −0.237 e Å−3 | 0.176 and −0.128 e Å−3 |
The X-ray structures of 1B, 2B and 3B showed dimers formed via C–H⋯O ‘head to head’ (Table 2, Fig. S14 in the ESI†) orientations of their respective molecules. Molecules of 2B differed by their slightly slipped C–H⋯O interactions. 2B is positioned in the vicinity of nine neighbouring molecules (Fig. S15 in the ESI†) via C–H⋯O and π–π stacking interactions, whereas 3B showed intermolecular hydrogen bonding to three neighbouring molecules via C–H⋯O interactions. Fig. 2 shows the molecular packing of 2B and 3B down the b and a-axis respectively. A polymeric sheet of dimer molecules formed via C(3)–H(3)⋯O(5) interactions in both the compounds, in addition the methyl group –C(13)H(13) took part in C–H⋯O interactions and slipped π–π stacking is observed for the 2B molecules (Fig. S16 in the ESI†). π–π stacking interactions are absent in compounds 1B and 3B. Compound 2B differs with respect to π–π stacking interactions from the rest of the compounds 1B and 3B, and this aspect could be reflected in the anticancer activity of these compounds.
| Compound | D–H⋯A | D–H (Å) | H⋯A (Å) | D⋯A (Å) | ∠D–H⋯A (°) | |
|---|---|---|---|---|---|---|
| a (i) −1/2 + x, 1.5 − y, −1/2 + z; (ii) −1/2 + x, 2.5 − y, −1/2 + z; (iii) 1.5 − x, −1/2 + y, 1/2 − z; (iv) 2 − x, 1 − y, −z; (v) 1 − x, −y, 2 − z. | ||||||
| 2B | C(13)–H(13A)⋯O(5)(i) | 0.980(1) | 2.672(1) | 3.299(1) | 122.11(8) | |
| C(13)–H(13C)⋯O(5)(ii) | 0.980(1) | 2.654(1) | 3.626(3) | 171.52(8) | ||
| C(3)–H(3)⋯O(5)(iii) | 0.951(1) | 2.586(1) | 3.437(1) | 149.13(6) | ||
| C(8)–H(8)⋯O(7)(iv) | 0.951(1) | 2.700(1) | 3.640(3) | 169.92(6) | ||
| 3B | C(3)–(H3)⋯O(5)(v) | 0.931(2) | 2.677(1) | 3.362(2) | 131.0(1) | |
| C(8)–(H8)⋯O(5)(v) | 0.930(2) | 2.675(1) | 3.564(2) | 160.2(1) | ||
| C(6)–(H6)⋯O(7)(v) | 0.930(1) | 2.657(1) | 3.560(2) | 163.9(1) | ||
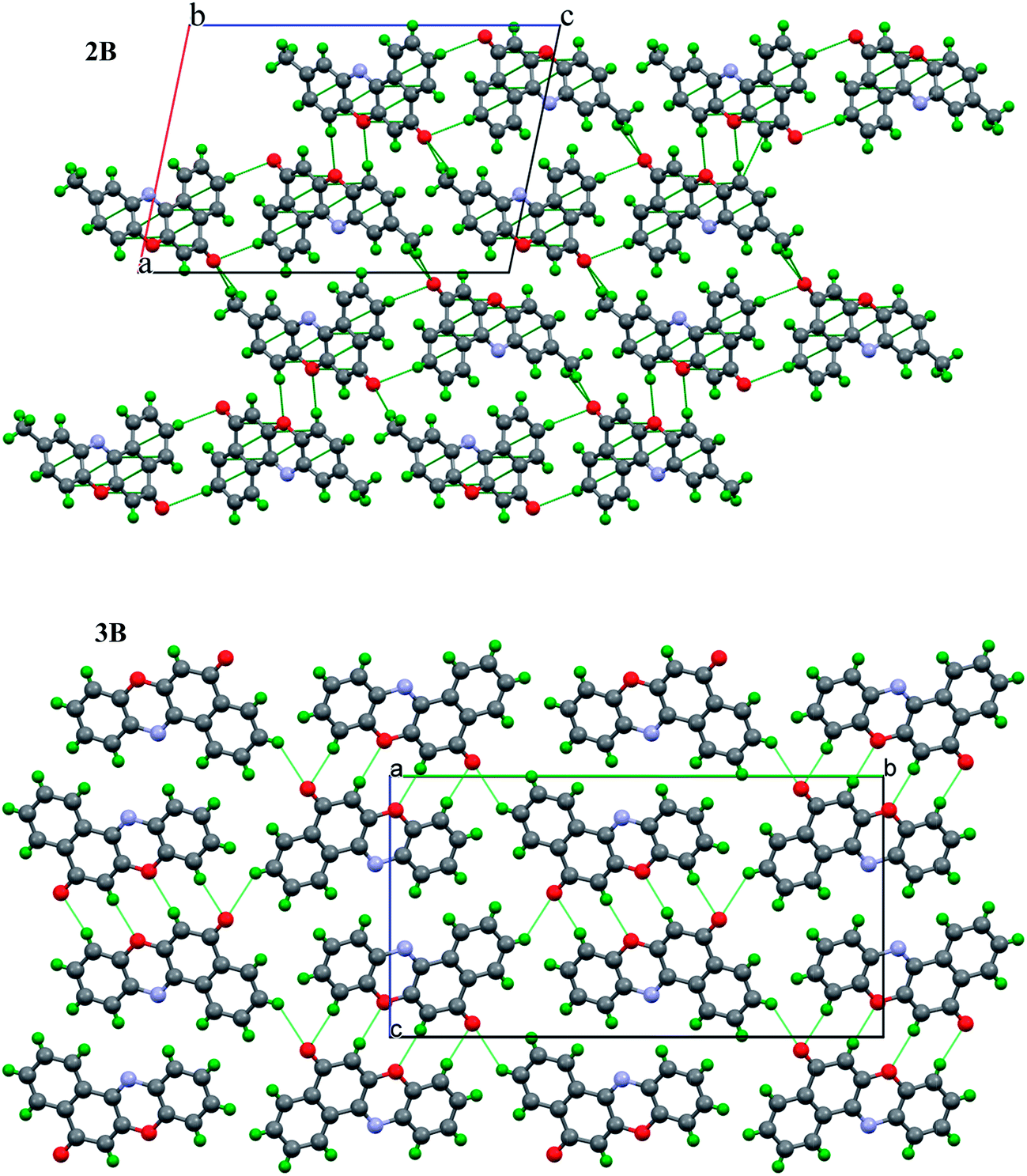 | ||
| Fig. 2 Molecular packing of 2B down the ‘b’-axis (top) and 3B down the ‘a’-axis (bottom). | ||
Computational studies
The structures of 1B, 2B and 3B obtained using M06-2x based density functional theory are depicted in Fig. S17 (in the ESI†). Selected bond distances (in Å) are given along with them. The structures obtained from the dispersion corrected density functional theory are in consonant with single crystal data derived from the X-ray diffraction experiments. To gain deeper insight into the charge distributions within these systems, the molecular electrostatic potential (MESP) was computed within the same framework of theory. The MESP, V(r), results from the balance of bare nuclear and electronic contributions brings about the effective electron-rich regions in the molecule. The MESP isosurfaces with V = −52.5 kJ mol−1 are compared in Fig. 3. As may readily be inferred, the electron-rich regions are located near the carbonyl oxygens in 1B–3B. Furthermore the softness parameters (η) were computed from the difference of the HOMO (highest occupied molecular orbital) and LUMO (lowest unoccupied molecular orbital) energies. The calculated η value for 2B turns out to be 0.109 eV, compared to that of 0.087 eV for 1B and 0.015 eV for 3B.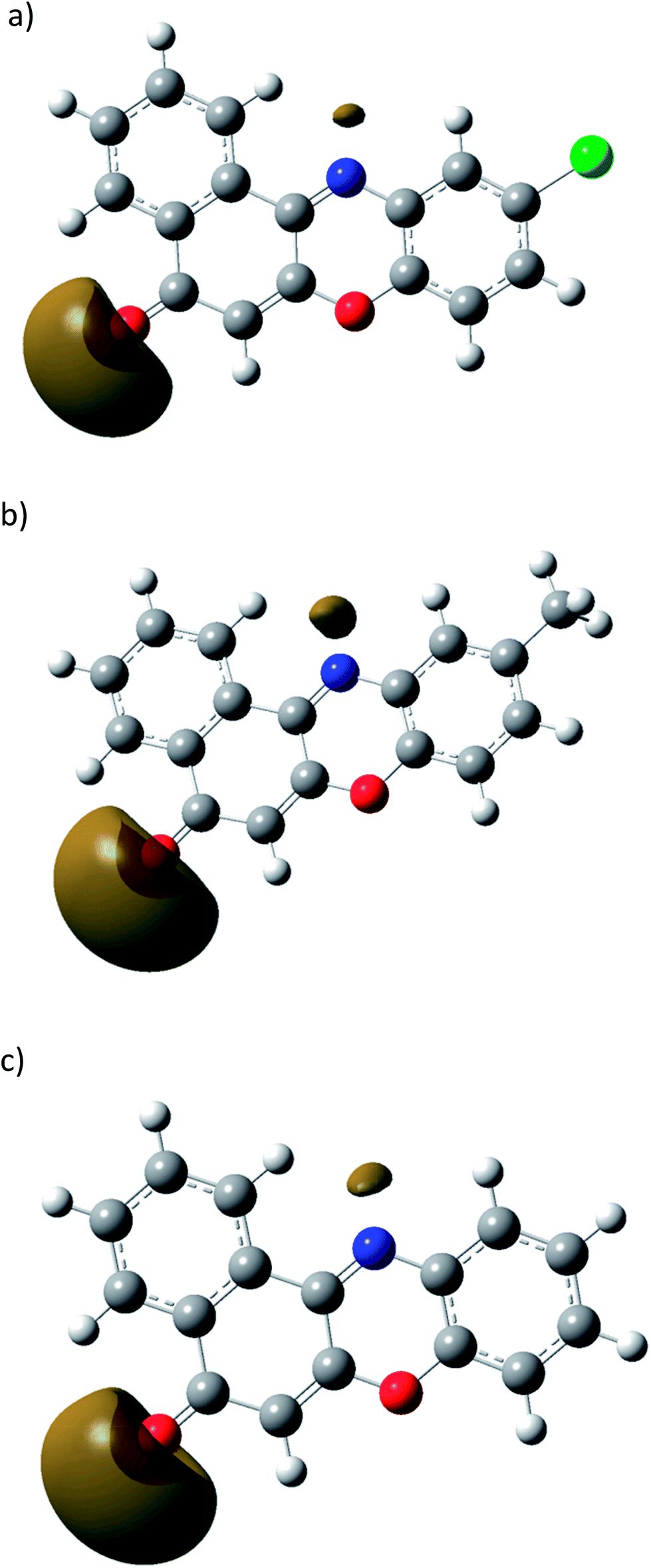 | ||
| Fig. 3 Molecular electrostatic potential surface (0.02 a.u isosurface) for (a) 1B (b) 2B and (c) 3B. | ||
Antiproliferative activities of 1B, 2B and 3B, observed only in BRAF V600E colon cancer cells
The cell viability upon treatment with the three compounds at various concentrations of COLO205 (V600E BRAF, malignant) and HEK293T (wild type BRAF, transformed non-malignant) showed that compounds 2B and 3B posses an excellent anti-proliferative activity specifically for COLO205, with IC50 values of 13 μM and 9 μM, while for compound 1B antiproliferative activity was observed only at concentrations above 50 μM (Fig. 5). No or negligible effects were observed for the HEK293T cell line even when treated with doses as high as 100 μM (Fig. 4). These data demonstrate that the compounds were able to induce cytotoxicity selectively in the BRAF mutated colon cancerous cells and that the very significant activity of compounds 2B and 3B in comparison to 1B indicates a strong structure activity relationship. | ||
| Fig. 4 Dose-dependent cell viability data for compounds 1B, 2B and 3B upon treatment of COLO205 and HEK293T cells for 72 h. Results (n = 3) are expressed as the mean ± SD and IC50 doses were determined for the bioactive compounds. The doses that were used for further studies of the IC50 value for the bioactive compounds, or 50 μM for the inactive compounds, are shown in the panel below. Doxorubicin (DOX) was used as a positive control. | ||
Compounds 1B, 2B and 3B, with a substitution for –R of Cl, CH3 and H, are expected to be cell permeable with a logP value of 2.87, 2.79 and 2.33 respectively. As these compounds are innately fluorescent, flow cytometry and fluorescence microscopy were performed to study their cell permeability, retention and localization. From the analysis, only 2B and 3B were bioactive in COLO205 cells and the others were inactive. Therefore, for all the following studies using COLO205, 1B, 2B and 3B were used at 50 μM (high dose), 13 μM (IC50) and 9 μM (IC50) concentration respectively and for HEK293T all the compounds were used at a high dose of 50 μM.
Comparative cell permeability of 1B, 2B and 3B
The excitation and emission wavelengths of all three fluorescent compounds [1B: Ex-475 nm/Em-535 nm, 2B: Ex-480 nm/Em-538 nm, 3B: Ex-470 nm/Em-540 nm] were compatible for fluorescence microscopy and flow cytometry studies. From the fluorescence microscopy studies, a green fluorescence was detected following 15 min incubation with the three compounds independently in both cell lines indicating cellular uptake in all cases. From DAPI stained nuclei, cytoplasmic accumulation of the uptaken compounds was confirmed for all three compounds in both the cell lines (Fig. 5).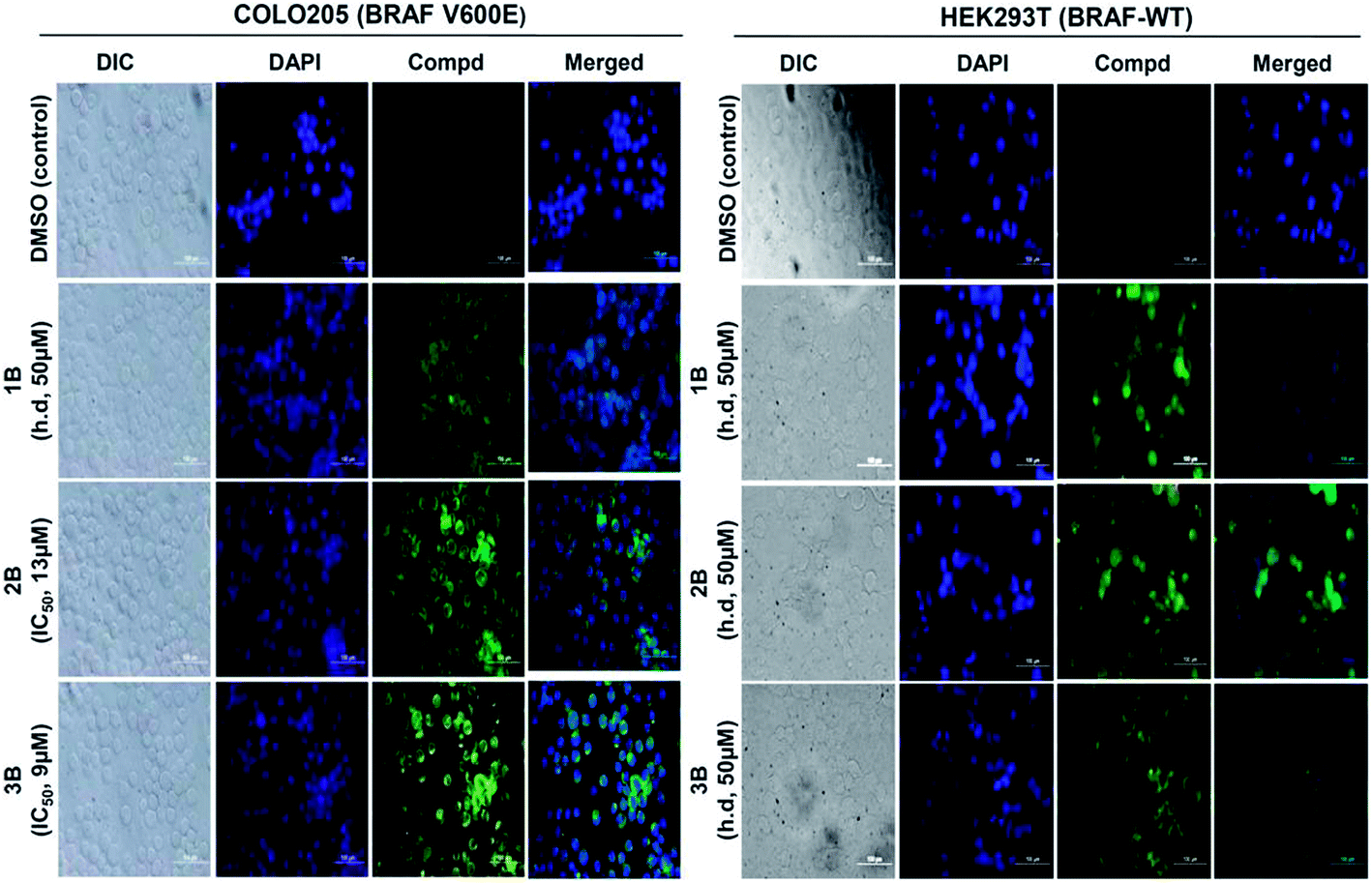 | ||
| Fig. 5 Cell permeability studies for compounds 1B, 2B and 3B using fluorescence microscopy within 15 min post exposure. COLO205 and HEK293T cells were incubated with the compounds (1B, 2B and 3B) at the above-mentioned concentrations for 15 min. Cytosolic accumulation of the compounds (green fluorescence) was confirmed when the nucleus was counter-stained (blue color) with DAPI. The cells were either treated with the IC50 concentrations or a high dose of 50 μM for the inactive compounds. | ||
Under similar dose conditions, the cellular uptake was quantified using flow cytometry following incubation with the compounds under study for 15 min, 2 h and 24 h. From the results a differential cellular uptake was observed for the compounds in both cell lines at different time points. 1B treated at a high dose of 50 μM still had much less uptake for both cell lines. 2B and 3B treated on HEK293T at a dose of 50 μM, that is approximately five times higher than the IC50 concentration used for COLO205, showed a relatively higher uptake of 2B in HEK293T in comparison to the COLO205 treated at IC50. But for compound 3B, the uptake was almost the same at all time-points whether it was COLO205 treated at IC50 or HEK293T treated at 50 μM (Fig. 6). The cell viability assay and cellular uptake studies show the poor uptake of 1B and an inability to reach the intracellular cytotoxic threshold concentration as a possible reason for its less potent antiproliferative activity. For 2B and 3B the treatment doses and cellular uptakes in COLO205 were low in comparison to HEK293T, but still these two compounds were very cell permeable and able to effectively inhibit proliferation of COLO205 by 50%. Taking together these results we concluded that 2B and 3B are highly selective for malignant COLO205 cells and that the cellular environment under the V600E BRAF mutated cancerous conditions is responsible for their antiproliferative activity. Furthermore, in order to understand in more detail the mechanistic activity of this selective cell death, studies for cell cycle arrest and apoptotic activity were performed for these compounds.
 | ||
| Fig. 6 Flow cytometric analysis for the cellular uptake of the compounds (at 15 min, 2 h and 24 h post treatment) using COLO205 and HEK293T. The extent of the cellular uptake in terms of mean fluorescence intensity (MFI) shows that the HEK293T cells were more refractory towards all three compounds even at a high dose (h.d, 50 μM) compared to COLO205, where compound 1B was treated at a high dose of 50 μM and both 2B and 3B were treated at their IC50 doses. | ||
1B, 2B and 3B induced cell cycle arrest in BRAF V600E colon cancer cells
Post 24 h exposure with the compounds, cell cycle analysis using flow cytometry showed a significant number of cells arrested at the G0/G1 phase only in the COLO205 cells treated with 1B, 2B and 3B (Fig. 7). Although the HEK293T cells were treated at high doses, their cell cycle progression seems unaffected post 24 h treatment (Fig. 7). Apart from the cell cycle arrest at the G0/G1 phase, a concomitant accumulation of the sub G1 population was observed that increased after 24 h treatment which may be either due to apoptosis or necrosis. So further experiments were performed to elucidate the preferred cell death pathway (apoptosis or necrosis) induced by these compounds.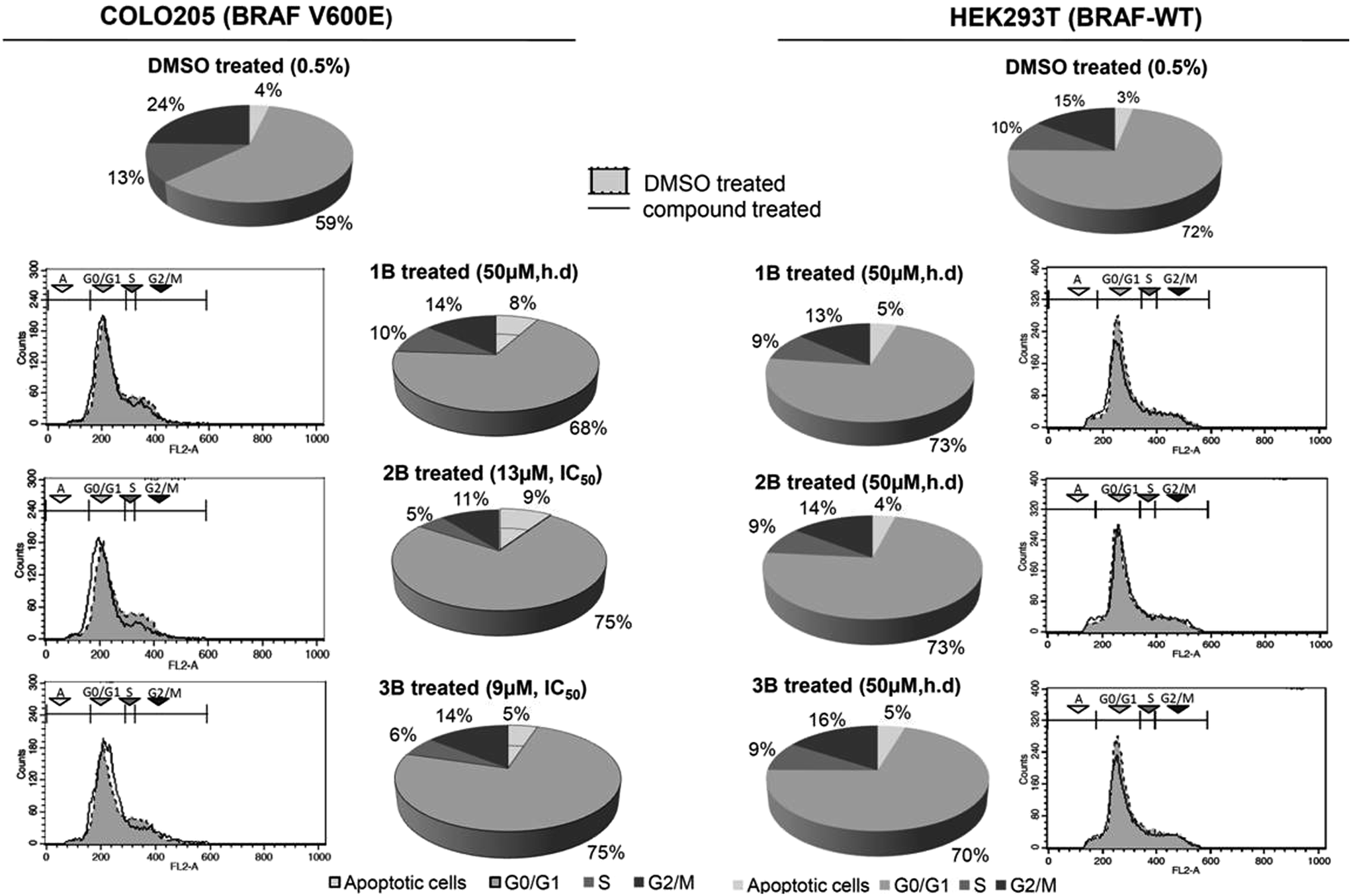 | ||
| Fig. 7 Propidium iodide staining for cell cycle studies using flow cytometry. G0/G1 phase arrest in the cell cycle was observed in the COLO205 cells (left panel). Compounds 2B and 3B were able to show equivalent activity at concentrations 5 times lower than that of compound 1B. No cell cycle arrest was observed in the HEK293T cells (right panel) with the compounds even when treated at a high dose (h.d.) of 50 μM. | ||
1B, 2B and 3B induce apoptosis in V600E BRAF colon cancer cells
Altered or deregulated apoptosis is reported in almost all tumor cells with a significant role in promoting malignancy. To assess the apoptosis promoting ability of these compounds at the determined doses we studied the appearance of various apoptotic hallmarks (the increment of annexin V positive cells, chromosomal DNA fragmentation and active or cleaved caspase-3 accumulation) in the treated cells at various time points after treatment.Within 2 h post treatment for the COLO205 cells, the compounds showed an increased amount of annexin-V positive cells due to alteration of the membrane asymmetry and as an indicator for early markers of apoptosis (Fig. 8). From the 24 h treatment, an increase in the percentage of annexin-V positive and 7AAD/annexin-V double positive cells was observed only in the COLO205 cells, indicating severe cell death (data not shown). But the treated HEK293T cells remained unaffected even after 24 h treatment at high doses.
 | ||
| Fig. 8 Annexin-V assay for the assessment of apoptotic activity in 1B, 2B and 3B treated COLO205 and HEK293T cells. Apoptosis was determined after treating both cell lines with compounds 1B, 2B and 3B followed by staining with annexin-V/7-AAD after 2 h (top). Flow cytometry profiles (scatter plots, top panel) represent annexin-V-PE staining along the x-axis (FL2-H) and 7-AAD along the y-axis (FL3-H). Histograms for early apoptosis after 2 h and 24 h are expressed as a percentage for each experimental condition (lower panel). | ||
This increase in 7AAD/annexin-V double positive cells due to cell death was further confirmed using a DNA fragmentation assay, a late marker of apoptosis, to establish the compounds as potential pro-apoptotic agents.
DNA fragmentation, as an inference for late markers of apoptosis, following 48 h of treatment was only observed for COLO205, even at very low doses (except for 1B). No fragmented chromosomal DNA (apoptosis) or smear (necrosis) was observed for the treated HEK293T cell lines. These results confirmed that the tested compounds are potent apoptosis inducers selective for malignant BRAF V600E colon cancer cells and that non-malignant wild type BRAF cells will be unaffected by their treatment (Fig. 9).
 | ||
| Fig. 9 Compounds 1B, 2B and 3B induced DNA fragmentation only in COLO205 (BRAF V600E) colon cancer cells. After a 48 h treatment of both the HEK293T and COLO205 cells with compounds 1B, 2B and 3B at the indicated doses (high dose of 50 μM for inactive and IC50 dose for active compounds), the chromosomal DNA was extracted. On a 1.8% agarose gel, a DNA laddering profile was observed for the COLO205 (BRAF V600E) cell lines when treated with compounds 1B, 2B and 3B for 48 h, indicating stronger apoptotic activity. This was not observed in the HEK293T cells where the BRAF is wild-type. | ||
1B, 2B and 3B induced caspase-dependent apoptosis in V600E colon cancer cells
To further investigate the mode of apoptosis in COLO205 cells, a western blot was performed to detect active caspase-3 (cleaved) following 12 h treatment with the compounds. The appearance of cleaved caspase-3 is a hall mark for caspase-dependent apoptosis. In our experiment, accumulation of the distinct, active as well as cleaved form of caspase-3 was observed only in the 1B, 2B and 3B treated COLO205 cell lysates while the active form of this executioner pro-apoptotic protein was absent in the 1B, 2B and 3B treated HEK293T cell lysates (Fig. 10).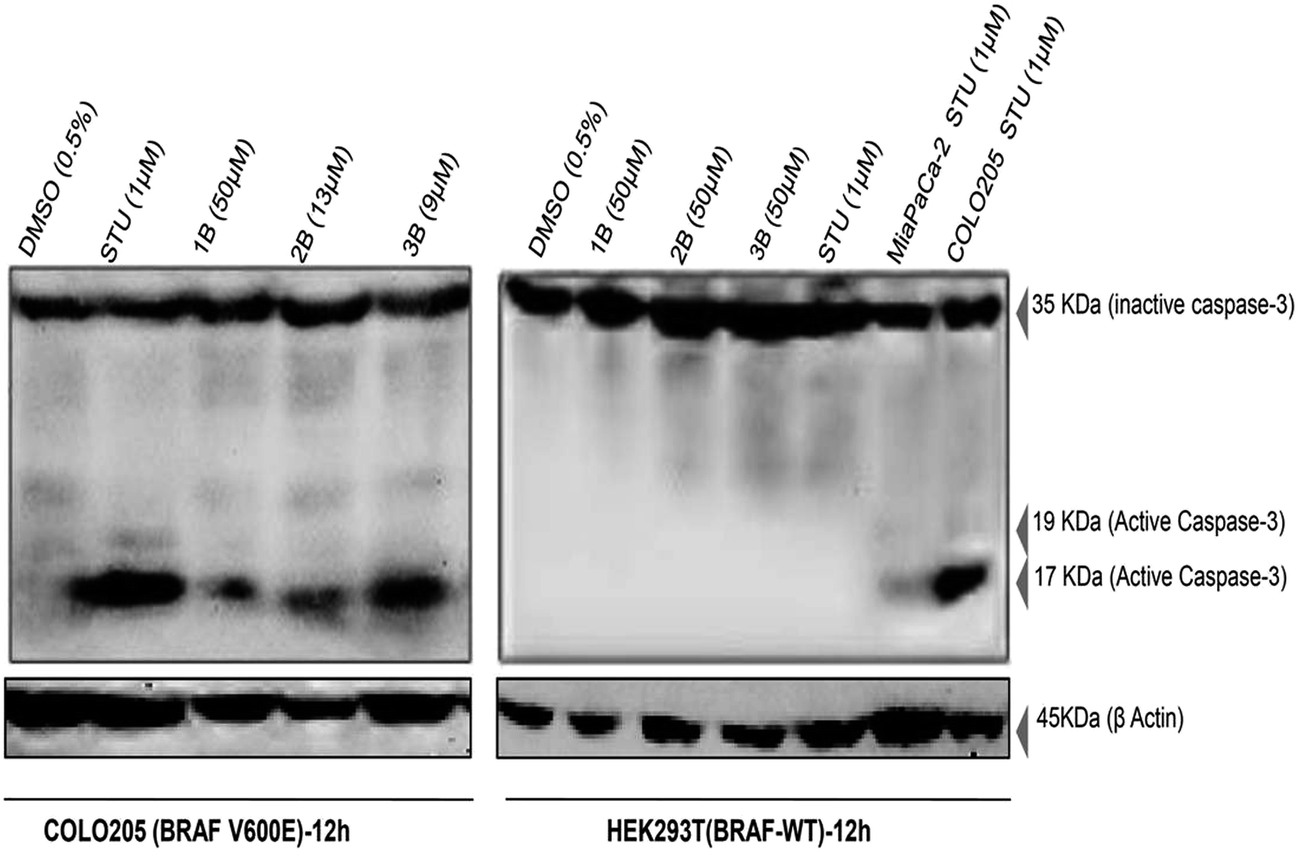 | ||
| Fig. 10 Compound 1B, 2B and 3B induced caspase-3 dependent apoptosis in COLO205 (BRAF V600E) cells. Caspase-3 activation by compounds 1B, 2B and 3B after 12 hours of treatment was detected (17 kDa band) with a primary antibody (anti-human caspase-3 at 1:1000 dilution) and anti-human β-actin at 1:1000 dilution used to detect β-actin that was used as a loading control. 1 μM staurosporine (STU) treated cell lysates were used as a positive control. | ||
From these experiments, we could conclusively state that 1B, 2B and 3B despite being cell permeable were selective in inducing cytotoxicity in BRAF-V600E colon cells by causing cell cycle arrest accompanied by a caspase-dependent apoptotic pathway.
Conclusions
In the era of personalized medicine, single treatment or individualized treatment strategies against BRAF mutated colon cancer and other adaptive resistant cancer cells are not available due to the harboring of different treatment refractory mutations. A small number of p.BRAF V600E targeted inhibitors (e.g. vemurafenib) are currently in different stages of clinical trials against melanomas but their efficacy is very low in similarly mutated colon cancer cells due to extremely poor clinical response. Moreover from recent clinical studies, many cases of acquired resistance to vemurafenib have also been reported.40In the present study, we report the cellular uptake of all three fluorescent compounds in both cell lines, but antiproliferative activity was observed in the BRAF V600E mutated colon cancer cell line, COLO205, when treated with 2B and 3B. Further detailed studies confirmed cell cycle arrest (G0/G1) and induction of caspase-dependent apoptosis to be the possible mechanism for the antiproliferative activity of 2B and 3B. Apart from the potent anti-proliferative activity, a high degree of specificity for BRAF mutated cancer was also observed with 2B and 3B, where both these compounds showed bioactivity only in the malignant colon cancer cells and had very minimal effect on the normal cells even at very high concentrations.
Another observation we report from our studies is that although one of the substituted benzo[α]phenoxazines 1B was inactive under such BRAF mutation conditions, it still did not show any cytotoxic activity towards the normal HEK293T cells at high concentration (same as was the case with 2B and 3B) unlike the other anticancer drugs (doxorubicin or staurosporine) that were used as positive controls. This is an important prerequisite while developing any chemotherapeutics, to address specificity and avoid cross reactivity making such a class of compounds attractive for further studies. More studies exploiting the fluorescent nature of these compounds or their prototypes can be useful in developing novel fluorophores for live imaging studies in animal models which would help in validating and establishing this class of compounds as potential anticancer agents against treatment refractory mutations and individualized treatments against colon cancer.
Experimental section
Materials for the chemical synthesis
All the chemicals used in the synthesis were of analytical grade. Lawsone (2-hydroxy-1,4-napthoquinone) and 2-aminophenol were obtained from Sigma-Aldrich and recrystallized from dry methanol before use. 4-Chloro-2-aminophenol and 4-methyl-2-aminophenol were obtained from Acros chemicals. The anhydrous methanol used in the synthesis was purified using a literature reported procedure.41Synthesis of 1B to 3B
Recrystallized 2-hydroxy-1,4-naphthoquinone (lawsone), 1 mM (174 mg), was dissolved in 20 ml of dry methanol. The solution was stirred for 15 min. The solids 4-chloro-2-aminophenol, 1 mM (143 mg), for 1B, 4-methyl-2-aminophenol, 1 mM (123 mg), for 2B, and 2-aminophenol, 1 mM (109 mg), for 3B, were dissolved in 10 ml of dry methanol, respectively. The aminophenol solutions were added dropwise into the solution of lawsone. The colour of the reaction mixture turned from yellow to brown. The mixture was stirred at room temperature (26 °C) for 24 h for 3B, and refluxed for 62 hours for 1B and 2B. The reaction was monitored using TLC (9.5:0.5; toluene:methanol). The dark red colour products obtained were filtered and washed with a small amount of methanol, then dried under vacuum and purified using column chromatography. A fluorescent yellow band was separated as the minor product for 1B to 3B with ∼10% yield, while a dark red band was separated as the major product (1A to 3A) by column chromatography (Scheme 3).
 | ||
| Scheme 3 General reaction scheme for the synthesis of 1B, 2B and 3B. | ||
FT-IR, elemental analysis, 1H, 13C and 2D gHSQCAD NMR, LC-MS, HR-MS, and cyclic voltammetry studies
The FT-IR spectra of the compounds were recorded between 4000–400 cm−1 as KBr pellets on a SHIMADZU FT 8400 spectrometer. The elemental analyses were performed using a Thermo Finnigan EA 1112 Flash series Elemental Analyzer. 1H, 13C and 2D gHSQCAD NMR spectra of 1B to 3B were recorded in CDCl3, using a Varian Mercury 500 MHz NMR spectrometer with TMS (tetramethylsilane) as a reference. UV-Vis spectra of the compounds were recorded using a SHIMADZU UV 1650 in methanol between 200 to 800 nm. HR-MS spectra were recorded using a Bruker Impact HD with an ESI source.Electrochemical measurements were performed with a CHI 6054E electrochemical analyser. A commercial Pt disc electrode (CHI Instruments, USA, 2 mm diameter), AgNO3 wire and Pt wire loop were used as the working, reference and counter electrodes respectively. After fixing the electrodes to the cell, 0.513 g of tetrabutylammonium perchlorate (100 mM in 15 ml solution) was transferred to the cell through high purity argon gas. The blank or control voltammograms were acquired using a tetrabutylammonium perchlorate–DMSO mixture prior to the measurements. The sample dispersed in small amount of solvent was injected into the cell for further measurements (analytic concentration of 5 mg/15 ml). At the end of each set of experiments the potentials were calibrated with respect to the normal hydrogen electrode (NHE) using ferrocene as an internal standard.
X-ray diffraction studies
An orange single crystal of 2B was coated with perfluoropolyether, picked up with nylon loops and mounted in the nitrogen cold stream of a Bruker APEX-II diffractometer. Graphite monochromated Mo-Kα radiation (λ = 0.71073 Å) from a Mo-target rotating-anode X-ray source was used. The final cell constants were obtained from the least squares fitting of several thousand strong reflections. The intensity data were corrected for absorption using the intensities of redundant reflections with the program SADABS.42 The structure was solved readily by direct methods and subsequent difference Fourier techniques. A Siemens ShelXTL43 software package was used for solving and drawing the structures, ShelXL9744 was used for the refinement. All the non-hydrogen atoms were anisotropically refined and the hydrogen atoms were placed at calculated positions and refined as riding atoms with isotropic displacement parameters.
Single crystals of 3B were grown by slow evaporation of methanol solution. Data was collected with a SMART APEX-II CCD using Mo-Kα radiation (λ = 0.7107 Å) to a maximum θ range of 25.00°. An orange coloured needle-like crystal of approximate size 0.38 × 0.09 × 0.04 mm3 was used for data collection. Crystal to detector distance was 5.00 cm, using 512 × 512 pixels per frame, an oscillation per frame of −0.5°, a maximum detector swing angle = −30.0°, a beam center = (260.2, 252.5), in plane spot width = 1.24, and multirun data acquisition. Total scans = 2, total frames = 657, exposure per frame = 10.0 s per frame, θ range = 0.66 to 25.00°, and completeness to θ of 23.97° is 99.6%. C16H9NO2, M = 247.24. Crystals belong to monoclinic space group P21/n, a = 3.91910(9), b = 23.3060(5) Å, c = 12.3580(3) Å, V = 1125.56(4) Å3, Z = 4, Dc = 1.459 g cm−3, μ (Mo-Kα) = 0.097 mm−1, 7783 reflections were measured, 1766 unique [I > 2σ(I)], the R value was 0.0350, and wR2 = 0.0901. Largest diff. peak and hole were 0.176 and −0.128 e Å−3. All the data were corrected for Lorentzian, polarisation and absorption effects. SHELX-97 (ShelxTL)43,44 was used for the structure solution and full matrix least squares refinement on F2. Hydrogen atoms were included in the refinement as per the riding model and refined. The data collection and refinement parameters are listed in Table 1.
Computational methodology
The optimized structures of 1B, 2B and 3B were obtained within the framework of M06-2x45,46 density functional theory using the Gaussian-09 program.47 An internally stored 6-31++G (d,p) basis set with the diffuse functions being added on the heavy atoms was employed.45–47 The structures were confirmed to be local minima on the potential energy surface through vibrational frequency calculations.Chemicals and reagents for the bioactivity studies
Stock solutions of 1B, 2B and 3B were prepared at a concentration of 20 mM in DMSO [MP Biomedicals, #196055] and stored at −20 °C. The chemicals and reagents used in the experiments, XTT (2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide) (#158788), PMS N-methyl dibenzopyrazine methylsulfate (#194595), doxorubicin (#159101) RNaseA (#0107680), proteinase K (#9398125), propidium iodide (#19545810), phenol:chloroform saturated solution, pH 6.7 (#154903), EDTA (#194822), Triton X-100 (Cat No. 194854), DAPI (#15757405) ethidium bromide (EtBr, #802511), polysorbate 20 (Tween-20, #103168), SDS (#102918), and protease inhibitor cocktail (#158837), were procured from MP Biomedicals. The buffers and media such as 10× PBS (#ML023), DMEM (#AL007A), RPMI (#AL162S), and foetal bovine serum (#RM9970) were purchased from HIMEDIA, while Tris–HCl (#T5941), Trizma base (#T1503), sodium chloride (#S3014) and methanol (#154903) were purchased from Sigma. Agarose for the agarose gel electrophoresis was procured from Conda (#8012). Antibodies and reagents for performing western blotting were purchased from Cell Signaling, primary rabbit anti-human caspase3 (#9665), anti-β-actin (#4970), secondary goat anti-rabbit HRP-IgG (#7074) and 20× lumiGLO® ECL reagent and 20× peroxide (#7003S). PVDF membrane (#1620177) and acrylamide/bisacrylamide solution 30% (29:1) (#1610156) were purchased from Bio-rad. A pre-stained protein ladder was bought from Fermentas (#SM0671) to perform the SDS-PAGE.
Cell lines and culture conditions
Both human malignant colon cancer cell line COLO205 (heterozygous for BRAF: c.1799T>A or p.V600E) and transformed, non-malignant human embryonic kidney cell line HEK293T (wild type BRAF and inactivated p53 by expressing SV40 large T antigen) were cultured in RPMI-1640 and DMEM growth medium respectively in an incubator maintained at 37 °C with a constant supply of 5% CO2. All media were supplemented with 10% heat-inactivated fetal bovine serum (FBS), 1% L-glutamine and 1% penicillin–streptomycin. During each experiment the final FBS concentration was maintained at 5% in all growth media.In vitro cell viability assay
A colorimetric assay using XTT was performed for both the cell lines (COLO205 and HEK293T) to study the effects of compounds 1B, 2B and 3B on the cell proliferation activity. Briefly, 100 μL of media containing 1 × 104 cells was placed into each well of a 96-well plate and incubated for a minimum of 8 h for cell adherence. Later, the cells were incubated independently with increasing concentrations (10−8 to 10−4 M) of all three test compounds for 72 h at 37 °C in a 5% CO2 incubator. Each experimental 96-well plate had cells similarly treated with doxorubicin which served as a positive control while cells treated with DMSO at a final concentration of 0.5% were used as the solvent control. Duplicate wells were used for each treatment. Post incubation, 50 μL of an assay mix consisting of XTT and PMS (49:1) was added to each well. After about 8 h incubation, the enzymatic activity of the mitochondrial dehydrogenase present in metabolically active cells cleaved the XTT releasing an orange formazan dye. The intensity of the coloured product formed was taken as a measure of the number of active cells present. A micro-plate reader (iMark, Biorad) was used to measure the intensity of the absorbance from the coloured product formed by setting the wavelength at 490 nm with a reference wavelength of 655 nm. The percentage cell viability was calculated using the following equation,| Cell viability (%) = (O.D sample/O.D DMSO treated control) × 100 |
Cellular permeability and localization studies using fluorescence microscopy
About 1 × 104 cells of COLO205 and HEK293T were grown on cover slips placed in each well of a 24-well plate containing the respective growth medium. The cells in each well were then treated with the fluorescent test compounds 1B, 2B and 3B at the required final concentration (either at IC50 or a high dose of 50 μM) and incubated for up to 24 h at 37 °C under a continuous flow of 5% CO2. Post incubation, the cells were washed twice with PBS and fixed with 2% paraformaldehyde. The cells were fixed and the nuclei were counterstained with DAPI. Whole cell images were captured at 40× magnification using a fluorescence microscope (Olympus) and applying appropriate wavelength filters.Quantitative cellular uptake studies using flow cytometry
About 1 × 106 COLO205 and HEK293T cells were incubated with test compounds 1B, 2B and 3B either at a final concentration of the IC50 or at 50 μM in growth media at 37 °C. Post treatment, the cells were harvested after time points of 15 min, 2 h and 24 h followed by washing with ice cold PBS. Finally, single cell suspensions were prepared in 500 μL of PBS to determine the mean fluorescence intensity at a compensated FL1 voltage by flow cytometry using a FACS-Calibur flow cytometer (BD Biosciences) and using excitation and emission wavelengths compatible with the same channel for individual compounds [1B: Ex-475 nm/Em-535 nm, 2B: Ex-480 nm/Em-538 nm, 3B: Ex-470 nm/Em-540 nm]. 5000 gated live events were taken from the forward vs. side scattering plots (ensuring similar sizes and granularity for both treated as well as untreated conditions), to analyse the extent of the compound uptake into the cells after each indicated time point. Histograms were obtained by comparing the mean fluorescence intensity of the test samples with that of the DMSO treated control samples.Cell cycle analysis
The effects of 1B, 2B and 3B on cell cycle progression were studied using flow cytometry (BD Biosciences). Briefly, post 24 h incubation with the test compounds at IC50 or 50 μM concentration with 106 cells per ml, the growth medium was discarded and the cells were harvested with ice cold PBS and fixed in 3% paraformaldehyde before staining with a mixture containing propidium iodide (PI), Triton X-100 and RNaseA. 5000 live gated events were acquired using a flow cytometer. Cell Quest Pro software was used to generate histograms based on the DNA content in each phase of the cell cycle.Annexin-V/7AAD assay
An annexin-V assay was carried out using flow cytometry to examine the apoptosis induction of COLO205 and HEK293T cells in response to treatment with the test compounds. Briefly, 1 × 106 live cells were seeded into 24-well plates containing complete growth medium in the absence or presence of the test compounds, treated at IC50 or 50 μM, and incubated at 37 °C under 5% CO2. After 2 h and 24 h of treatment, the cells were harvested and washed twice with ice cold PBS. Single cell suspensions were prepared using 1X binding buffer (BD biosciences). Staining was performed as per the protocol mentioned in the user’s guide of the annexin-V apoptosis detection kit (BD biosciences #559763). The gated population obtained from the scatter plots and the cells undergoing apoptosis upon compound treatment were determined and represented in terms of percentage by comparing the test samples with the DMSO treated solvent control.DNA fragmentation studies
COLO205 and HEK293T cells at a density of 106 cells per ml were sown into 65 mm dishes containing the respective growth medium and incubated with the test compounds at IC50 or 50 μM concentration for 48 h at 37 °C under 5% CO2. 0.5% DMSO treated cells were used as a solvent control. The cells were lysed in lysis buffer containing 10 mM Tris–HCl (pH 8), 150 mM NaCl, 5 mM EDTA and 0.5% Triton X-100. The lysates were vortexed and incubated with 0.1 mg ml−1 RNaseA for 1 h at 37 °C followed by proteinase K (0.1 mg ml−1) treatment for another 2 h under the same conditions. Later the lysate was extracted for its chromosomal DNA using a phenol/chloroform/isoamyl alcohol mixture (25:24:1). The quality of extracted DNA was assessed using NanoDrop UV/Vis spectrophotometer (Thermo scientific) with a 260/280 absorbance ratio of 1.8–2.0. Equal amounts (in terms of DNA content) of the individual samples were loaded and electrophoresed on 1.8% agarose gel containing 0.1 μg ml−1 ethidium bromide to observe any fragmented chromosomal DNA.
Caspase-3 activation studies
About 1 × 106 COLO205 and HEK293T cells were sown into 35 mm dishes in the presence or absence of the test compounds and incubated for 12 h at 37 °C under 5% CO2. Post treatment, the samples were harvested with ice cold PBS and simultaneously lysed with 50 mM Tris buffer pH 8.0, 150 mM NaCl and 1% Triton X-100 along with a cocktail of protease inhibitors and the total protein concentration was measured at 280 nm with a nanodrop spectrophotometer (Thermo scientific). Later each sample was boiled with an equal amount of Laemmli sample buffer (4% sodium dodecyl sulfate (SDS), 10% β-mercaptoethanol, 20% glycerol, 0.004% bromophenol blue, 0.125 M Tris–Cl pH-6.8). Along with a pre-stained protein loading marker, 20 μL of the samples containing equal amounts of proteins were separated electrophoretically in 10% SDS-PAGE. Upon proper separation of the bands as observed from the marker, the resolved proteins were electroblotted from the gel to a methanol pre-activated PVDF membrane using a semidry blotter (Biorad). The protein transferred PVDF membranes were incubated with a blocking buffer (5% skimmed milk in TBST) for 1 h at room temperature on a rocker. The membranes were then incubated overnight with a primary antibody (anti-human caspase-3 and β-actin), both at 1:1000 dilutions. Following incubation with the primary antibody, the membranes were washed twice with TBST (Tween-20 in Tris-buffered saline) and once with TBS (without Tween-20) before incubation with the secondary anti-rabbit HRP–IgG (goat) for the next 1 h at room temperature. Chemiluminescence was detected under Gel Doc-XRS (Biorad) upon addition of an enhanced chemiluminescence ECL kit following the manufacturer’s instructions.
Statistical analysis
All experiments were performed in triplicate and the results are expressed as mean ± standard deviation, unless otherwise indicated.Acknowledgements
SP is thankful to NISER for the institutional fellowship. SSG and VBK are thankful to the RGYI scheme (BT/PR6565/GBD/27/456/2012) of the Department of Biotechnology, New Delhi, India. Thanks go to Rishikesh Patil for the NMR experiments.References
- J. A. McCubrey, L. S. Steelman, W. H. Chappell, S. L. Abrams, R. A. Franklin, G. Montalto, M. Cervello, M. Libra, S. Candido, G. Malaponte, M. C. Mazzarino, P. Fagone, F. Nicoletti, J. Basecke, S. Mijatovic, D. Maksimovic-Ivanic, M. Milella, A. Tafuri, F. Chiarini, C. Evangelisti, L. Cocco and A. M. Martelli, Oncotarget, 2012, 3, 1068–1111 CrossRef
.
- J. A. McCubrey, L. S. Steelman, W. H. Chappell, S. L. Abrams, G. Montalto, M. Cervello, F. Nicoletti, P. Fagone, G. Malaponte, M. C. Mazzarino, S. Candido, M. Libra, J. Basecke, S. Mijatovic, D. Maksimovic-Ivanic, M. Milella, A. Tafuri, L. Cocco, C. Evangelisti, F. Chiarini and A. M. Martelli, Oncotarget, 2012, 3, 954–987 CrossRef
- M. Marshall, Mol. Reprod. Dev., 1995, 42, 493–499 CrossRef CAS PubMed
- K. P. Hoeflich, D. C. Gray, M. T. Eby, J. Y. Tien, L. Wong, J. Bower, A. Gogineni, J. Zha, M. J. Cole, H. M. Stern, L. J. Murray, D. P. Davis and S. Seshagiri, Cancer Res., 2006, 66, 999–1006 CrossRef CAS PubMed
- A. Preto, J. Figueiredo, S. Velho, A. S. Ribeiro, P. Soares, C. Oliveira and R. Seruca, J. Pathol., 2008, 214, 320–327 CrossRef CAS PubMed
- S. E. Baldus, K. L. Schaefer, R. Engers, D. Hartleb, N. H. Stoecklein and H. E. Gabbert, Clin. Cancer Res., 2010, 16, 790–799 CrossRef CAS PubMed
- M. Beeram, A. Patnaik and E. K. Rowinsky, J. Clin. Oncol., 2005, 23, 6771–6790 CrossRef CAS PubMed
- R. Marais, Y. Light, H. F. Paterson, C. S. Mason and C. J. Marshall, J. Biol. Chem., 1997, 272, 4378–4383 CrossRef CAS PubMed
- J. Avruch, X. F. Zhang and J. M. Kyriakis, Trends Biochem. Sci., 1994, 19, 279–283 CrossRef CAS
- C. Wellbrock, M. Karasarides and R. Marais, Nat. Rev. Mol. Cell Biol., 2004, 5, 875–885 CrossRef CAS PubMed
- W. De Roock, B. Claes, D. Bernasconi, J. De Schutter, B. Biesmans, G. Fountzilas, K. T. Kalogeras, V. Kotoula, D. Papamichael, P. Laurent-Puig, F. Penault-Llorca, P. Rougier, B. Vincenzi, D. Santini, G. Tonini, F. Cappuzzo, M. Frattini, F. Molinari, P. Saletti, S. De Dosso, M. Martini, A. Bardelli, S. Siena, A. Sartore-Bianchi, J. Tabernero, T. Macarulla, F. Di Fiore, A. O. Gangloff, F. Ciardiello, P. Pfeiffer, C. Qvortrup, T. P. Hansen, E. Van Cutsem, H. Piessevaux, D. Lambrechts, M. Delorenzi and S. Tejpar, Lancet Oncol., 2010, 11, 753–762 CrossRef CAS
- A. S. Sameer, Frontiers in Oncology, 2013, 3, 114 CrossRef PubMed
- A. Thiel and A. Ristimaki, Frontiers in Oncology, 2013, 3, 281 CrossRef PubMed
- S. G. Sharma and M. L. Gulley, Arch. Pathol. Lab. Med., 2010, 134, 1225–1228 CAS
- R. Dienstmann and J. Tabernero, Anti-Cancer Agents Med. Chem., 2011, 11, 285–295 CrossRef CAS
- R. B. Corcoran, H. Ebi, A. B. Turke, E. M. Coffee, M. Nishino, A. P. Cogdill, R. D. Brown, P. Della Pelle, D. Dias-Santagata, K. E. Hung, K. T. Flaherty, A. Piris, J. A. Wargo, J. Settleman, M. Mino-Kenudson and J. A. Engelman, Cancer Discovery, 2012, 2, 227–235 CrossRef CAS PubMed
- H. Yang, B. Higgins, K. Kolinsky, K. Packman, W. D. Bradley, R. J. Lee, K. Schostack, M. E. Simcox, S. Kopetz, D. Heimbrook, B. Lestini, G. Bollag and F. Su, Cancer Res., 2012, 72, 779–789 CrossRef CAS PubMed
- H. Hao, V. M. Muniz-Medina, H. Mehta, N. E. Thomas, V. Khazak, C. J. Der and J. M. Shields, Mol. Cancer Ther., 2007, 6, 2220–2229 CrossRef CAS PubMed
- M. S. Reimers, E. C. Zeestraten, P. J. Kuppen, G. J. Liefers and C. J. van de Velde, Gastroenterology Report, 2013, 1, 166–183 CrossRef PubMed
- Z. X. Yuan, X. Y. Wang, Q. Y. Qin, D. F. Chen, Q. H. Zhong, L. Wang and J. P. Wang, PLoS One, 2013, 8, e65995 CAS
- A. Sartore-Bianchi, K. Bencardino, A. Cassingena, F. Venturini, C. Funaioli, T. Cipani, A. Amatu, L. Pietrogiovanna, R. Schiavo, F. Di Nicolantonio, S. Artale, A. Bardelli and S. Siena, Cancer Treat. Rev., 2010, 36(suppl. 3), S1–S5 CrossRef CAS
- A. S. Little, K. Balmanno, M. J. Sale, S. Newman, J. R. Dry, M. Hampson, P. A. Edwards, P. D. Smith and S. J. Cook, Sci. Signaling, 2011, 4, ra17 CrossRef PubMed
- A. Prahallad, C. Sun, S. Huang, F. Di Nicolantonio, R. Salazar, D. Zecchin, R. L. Beijersbergen, A. Bardelli and R. Bernards, Nature, 2012, 483, 100–103 CrossRef CAS PubMed
- K. Cichowski and P. A. Janne, Nature, 2010, 464, 358–359 CrossRef CAS PubMed
- S. Whittaker, R. Kirk, R. Hayward, A. Zambon, A. Viros, N. Cantarino, A. Affolter, A. Nourry, D. Niculescu-Duvaz, C. Springer and R. Marais, Sci. Transl. Med., 2010, 2, 35–41 Search PubMed
- C. Bokemeyer, E. Van Cutsem, P. Rougier, F. Ciardiello, S. Heeger, M. Schlichting, I. Celik and C. H. Kohne, Eur. J. Cancer, 2012, 48, 1466–1475 CrossRef CAS PubMed
- L. De Mattos-Arruda, R. Dienstmann and J. Tabernero, Clin. Colorectal Cancer, 2011, 10, 279–289 CrossRef CAS PubMed
- S. Ogino, K. Shima, J. A. Meyerhardt, N. J. McCleary, K. Ng, D. Hollis, L. B. Saltz, R. J. Mayer, P. Schaefer, R. Whittom, A. Hantel, A. B. Benson 3rd, D. Spiegelman, R. M. Goldberg, M. M. Bertagnolli and C. S. Fuchs, Clin. Cancer Res., 2012, 18, 890–900 CrossRef CAS PubMed
- R. S. Lo, Cell Res., 2012, 22, 945–947 CrossRef CAS PubMed
- S. D. Verma, N. Pal, M. K. Singh, H. Shweta, M. F. Khan and S. Sen, Anal. Chem., 2012, 84, 7218–7226 CrossRef CAS PubMed
- K. I. McLuckie, Z. A. Waller, D. A. Sanders, D. Alves, R. Rodriguez, J. Dash, G. J. McKenzie, A. R. Venkitaraman and S. Balasubramanian, J. Am. Chem. Soc., 2011, 133, 2658–2663 CrossRef CAS PubMed
- D. Chadar, S. S. Rao, A. Khan, S. P. Gejji, K. S. Bhat, T. Weyhermüller and S. Salunke-Gawali, RSC Adv., 2015, 5, 57917–57929 RSC
- L. Cincotta, J. W. Foley and A. H. Cincotta, Cancer Res., 1993, 53, 2571–2580 CAS
- J. Jose, A. Loudet, Y. Ueno, R. Barhoumi, R. C. Burghardt and K. Burgess, Org. Biomol. Chem., 2010, 8, 2052–2059 CAS
- R. Straussman, T. Morikawa, K. Shee, M. Barzily-Rokni, Z. R. Qian, J. Du, A. Davis, M. M. Mongare, J. Gould, D. T. Frederick, Z. A. Cooper, P. B. Chapman, D. B. Solit, A. Ribas, R. S. Lo, K. T. Flaherty, S. Ogino, J. A. Wargo and T. R. Golub, Nature, 2012, 487, 500–504 CrossRef CAS PubMed
- M. J. Sale and S. J. Cook, Biochem. J., 2013, 450, 285–294 CrossRef CAS PubMed
- L. Kathawate, P. V. Joshi, T. K. Dash, S. Pal, M. Nikalje, T. Weyhermüller, V. G. Puranik, V. B. Konkimalla and S. Salunke-Gawali, J. Mol. Struct., 2014, 1075, 397–405 CrossRef CAS PubMed
- D. Chadar, M. Camilles, R. Patil, A. Khan, T. Weyhermüller and S. Salunke-Gawali, J. Mol. Struct., 2015, 1086, 179–189 CrossRef CAS PubMed
- R. Patil, D. Chadar, D. Chaudhari, J. Peter, M. Nikalje, T. Weyhermüller and S. Salunke-Gawali, J. Mol. Struct., 2014, 1075, 345–351 CrossRef CAS PubMed
- N. Wagle, C. Emery, M. F. Berger, M. J. Davis, A. Sawyer, P. Pochanard, S. M. Kehoe, C. M. Johannessen, L. E. Macconaill, W. C. Hahn, M. Meyerson and L. A. Garraway, J. Clin. Oncol., 2011, 29, 3085–3096 Search PubMed.
-
D. D. Perrin, W. L. F. Armarego and D. R. Perrin, Purification of Laboratory Chemicals, Pergamon Press, Oxford, 1988, p. 260 Search PubMed
-
G. M. Sheldrick, SADABS, Bruker–Siemens Area Detector Absorption and Other Correction, Version 2008/1, University of Göttingen, Germany, 2006 Search PubMed
-
ShelXTL 6.14, Bruker AXS Inc., Madison, WI, USA, 2003 Search PubMed
-
G. M. Sheldrick, SHELX-97 Program for crystal structure solution and refinement, University of Göttingen, Germany, 1997 Search PubMed
- Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed
- Y. Zhao and D. G. Truhlar, J. Chem. Theory Comput., 2005, 1, 415–432 CrossRef CAS
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. J. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, R. Hasegawa, M. Ishida, T. Akajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian03, Revision, Gaussian, Inc., Pittsburgh, PA, 2003 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Characterization of 1B–3B, HR-MS figures (Fig. S1, S5 and S9), FT-IR figures (Fig. S2, S6 and S10), 1H and 13C NMR (Fig. S3, S7 and S11), 2D gHSQCAD (Fig. S4, S8 and S12), and cyclic voltammetry (Fig. S13). Crystallography figures (Fig. S14–S17) and crystallographic tables (Tables S1–S8). CCDC 1412950 and 1413927. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra14949e |
| This journal is © The Royal Society of Chemistry 2015 |