
Thin water films and magnesium hydroxide fiber growth†
Abstract
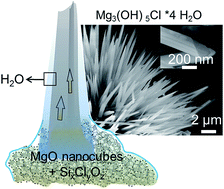
Thin films of water covering highly dispersed metal oxides can give rise to the spontaneous and spatially controllable growth of hydroxide fibers in ambient air. Knowledge about the underlying formation mechanisms is key to the rational development of metal oxide nanomaterials and associated microstructures. We used SiCl4 as a water free chlorine ion source for the surface functionalization of MgO nanocubes and explored their subsequent transformation into magnesium oxychloride Mg3(OH)5Cl·4H2O fibers upon contact with water vapor. Specifically we show how the temperature of the functionalization process and material dispersion control the reaction pathway that can lead to very different products like Mg(OH)2, MgCl2·6H2O, or Mg3(OH)5Cl·4H2O and delineate a reaction mechanism. Lessons to be learned from this unique route to form hydroxide fibers under ambient conditions can be applied to a variety of microstructural evolution processes that involve metastable solids and superficial water acting both as a reactant and as a reaction medium for the hydration of metal oxide particles.
 Please wait while we load your content...
Please wait while we load your content...

