
Li3V2(PO4)3 particles embedded in porous N-doped carbon as high-rate and long-life cathode material for Li-ion batteries†
Feifan Guoa,
Xiaoxin Zoua,
Kai-Xue Wangb,
Yipu Liua,
Feng Zhangc,
Yuanyuan Wua and
Guo-Dong Li*a
aState Key Laboratory of Inorganic Synthesis and Preparative Chemistry, College of Chemistry, Jilin University, 2699 Qianjin Street, Changchun 130012, China. E-mail: lgd@jlu.edu.cn
bSchool of Chemistry and Chemical Engineering, Shanghai Jiao Tong University, Shanghai, 200240, China
cSchool of Light Industry and Chemical Engineering, Dalian Polytechnic University, Dalian, 116034, China
First published on 10th September 2015
Abstract
Porous N-doped carbon stabilized Li3V2(PO4)3 particles are prepared by a modified sol–gel method. Both the in situ doping of nitrogen and the formation of porous structure can be attributed to the addition of dicyandiamide in the sol–gel process. The composite material exhibits a high discharge capacity of 114.7 mA h g−1 at 1 C in the voltage range of 3–4.3 V after 600 cycles. Even at a high rate of 5 C, the discharge capacity of 90.5 mA h g−1 is achieved after 600 cycles. The superior electrochemical performance mainly benefits from the porous and nitrogen doped carbon.
Introduction
Due to their high energy and high power density, rechargeable lithium ion batteries (LIBs) have been regarded as one of the most important energy storage systems for saving energy and reducing CO2 emission.1,2 It is well known that the electrochemical performance of cathode materials is the key issue for high performance LIBs.3–5 Up to now, LiCoO2 has been widely used as cathode material for commercial LIBs. However, it suffers from low practical capacity (140 mA h g−1), high cost, high toxicity and poor stability at high temperature.6–8 In order to overcome these shortcomings, some cathode materials based on polyanions phosphate, such as LiMPO4 (M = Fe, Co, Ni, Mn, …) and Li3M2(PO4)3 (M = V, Fe), have been extensively studied due to their good stability and/or high capacity.9–16 Among these materials, monoclinic Li3V2(PO4)3 (LVP) with a NASICON crystal structure has been considered as a promising cathode material for its robust three dimensional framework and a high theoretical capacity of 197 mA h g−1.15,17–19 Moreover, LVP also has some other significant features, including abundant raw materials, low toxicity, good ion mobility, high energy density, high operating voltage and good safety.18,20As a member of poly-anionic materials, LVP also exhibits a nature of low electronic conductivity (2.4 × 10−7 S cm−1) which limits its rate performance for practical applications.21 In order to eliminate these obstacles, numerous methods have been developed, including doping metal ions, decreasing the particles size and employing conductive layers. In particular, carbon coating is considered as a conventional and efficient way to improve the electronic conductivity of LVP.17–20,22–30 In this way, organic compounds are usually used to reduce V5+ to V3+ to form LVP under high temperature in inert atmosphere. And the residual carbon will be coated on the surface of the as-synthesized LVP at the same time. What's more, the carbon layer can also stabilize the LVP against the volume change during cycling. However, the thick dense carbon layer would hinder the migration of lithium ions into the interior of LVP, resulting in unsatisfied rate performance to some extent. Thus, porous carbon has been reported as a desired scaffold to overcome the disadvantages of dense carbon coating for LVP.31,32 It is believed that the pores in the carbon can reduce diffusion length of Li ions, leading to better rate capability.31,33 While, those porous carbon materials used in previous works are all prepared by rather complicated methods.34–36 Furthermore, non-metallic atom-doped carbon layer can further improve the electric conductivity and electrochemical activity of LVP.37,38 Nitrogen doping is one of the most attractive methods, which can create structural defects and increase the positive charge density of the carbon atoms adjacent to nitrogen, leading to the higher chemical reactivity and better electronic property for the composite material.37,39
Herein, we report a facile strategy for the preparation of Li3V2(PO4)3 particles embedded in porous N-doped carbon. In the synthetic process, dicyandiamide plays an important role in the formation of porous N-doped carbon. The as-synthesized products present superior electrochemical behaviour as the cathode material for LIB. The porous carbon in the composite could increase the interface between electrolyte and active materials, facilitating the diffusion of ion and electron in the material. And as a result, the cycling performance and the reversible capacity are increased greatly at high rate condition. In the voltage range of 3.0–4.3 V, the discharge capacities of the as-synthesized porous N-doped carbon stabilized Li3V2(PO4)3 material are retained 114.7, 110.5, 90.5 and 79.8 mA h g−1 after 600 cycles at 1, 2, 5 and 10 C, respectively.
Experimental
Materials synthesis
The porous nitrogen-doped carbon stabilized Li3V2(PO4)3 (LVP@PNC) composites were synthesized via a facile modified sol–gel method. For the synthesis of LVP@PNC, vanadium pentoxide (V2O5), oxalic acid (C2H2O4·2H2O), lithium nitrate (LiNO3), ammonium dihydrogen phosphate (NH4H2PO4), citric acid monohydrate (C6H8O7·H2O) and dicyandiamide (C2H4N4) were used as raw materials. In a typical synthesis, V2O5 and C2H2O4·2H2O in a stoichiometric ratio were added into 20 mL deionized water under magnetic stirring at 70 °C. In this process, V2O5 was turned to VO(C2O4). When a clear blue solution was formed, a mixture of stoichiometric amounts of LiNO3 and NH4H2PO4 were added to the above solution. After stirring for 10 minutes, 3.0 g of C6H8O7·H2O and 0.5 g of C2H4N4 were added and stirred for 5 h to form a gel. Afterwards, the gel was dried at 100 °C in air overnight. To prepare LVP@PNC, the dried precursor was calcined at 750 °C for 4 h at a rate of 5 °C min−1 under N2 atmosphere. For comparison, LVP@C, the sample without additional C2H4N4, was prepared at the same condition.Materials characterization
The structure of the LVP@PNC and LVP@C were identified by a Rigaku D/Max 2550 X-ray diffractometer (XRD) with Cu Kα radiation (λ = 1.5418 Å). The morphologies of the product were observed by a JEOL JSM 6700F electron scanning electron microscopy (SEM). The TEM and HRTEM images were taken on a Philips-FEI Tecnai G2S-Twin microscope equipped with a field emission gun operating at 200 kV (TEM). The N2 adsorption/desorption isotherms and Brunauer–Emmett–Teller surface areas of the composite were measured by a Micromeritics ASAP 2020M system. The Raman spectra were achieved on a Renishaw Raman system model 1000 spectrometer with an exciting line of 514.5 nm. The composition and the elemental state of the as-prepared compound was analyzed by an ESCALAB 250 X-ray photoelectron spectrometer with a monochromatized X-ray source (Al Kα hν = 1486.6 eV). The inductively coupled plasma (ICP) analysis was carried out on a Perkin-Elmer Optima 3300 DV ICP spectrometer. The thermogravimetric analysis (TGA) was performed on a NETZSCH STA 449C TG thermal analyser by heating the composites from room temperature to 750 °C in air. The infrared (IR) spectra were recorded on a Bruker IFS 66V/S FTIR spectrometer.Electrochemical measurements
The CR2016 coin-type cells were fabricated for investigating the electrochemical properties of LVP@PNC. The working electrode was prepared by mixing 80 wt% LVP@PNC, 10 wt% polyvinylidene fluoride (PVDF) and 10 wt% acetylene black (AB) in N-methyl-2-pyrrolidone (NMP). The mixture was stirred to form a uniform slurry which was then spread onto an aluminium foil and dried under vacuum at 120 °C for 12 h. The coin-type cell was assembled in an argon-filled glove box (Mikrouna), in which both the oxygen and moisture were less than 1 ppm. The electrolyte was 1 M LiPF6 dissolved in ethylene carbonate (EC)/dimethyl carbonate (DMC)/ethylene methyl carbonate (EMC) with a volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1. The lithium foil was used as anode. All the electrochemical performances of LVP@PNC were evaluated at room temperature (24 °C). The galvanostatic charge–discharge tests were performed between 3.0 V and 4.3 V on a LAND CT2001A cell testing system (Wuhan, China). The cyclic voltammetry (CV) was conducted in the cut-off voltage range of 3.0–4.3 V vs. Li/Li+ at a scan rate of 0.2 mV s−1 on a CHI660A electrochemical workstation (Chenhua, Shanghai).
:1:1. The lithium foil was used as anode. All the electrochemical performances of LVP@PNC were evaluated at room temperature (24 °C). The galvanostatic charge–discharge tests were performed between 3.0 V and 4.3 V on a LAND CT2001A cell testing system (Wuhan, China). The cyclic voltammetry (CV) was conducted in the cut-off voltage range of 3.0–4.3 V vs. Li/Li+ at a scan rate of 0.2 mV s−1 on a CHI660A electrochemical workstation (Chenhua, Shanghai).
Results and discussion
To identify the structure of the product, the XRD pattern of the LVP@PNC is shown in Fig. 1A. All of the diffraction peaks are well indexed to the monoclinic LVP (JCPDS no. 01-072-7074) with the space group P21/n.15 Furthermore, no obvious diffraction peaks related to graphite are observed in the XRD pattern due to the small amount of amorphous carbon. The structure of LVP is schematically shown in Fig. 1B, in which the 3D skeleton structure is composed of metal octahedron (VO6) and phosphorus tetrahedron (PO4). This 3D structure facilitates the reversible extraction and reinsertion of lithium ions within the host vanadium phosphate framework.28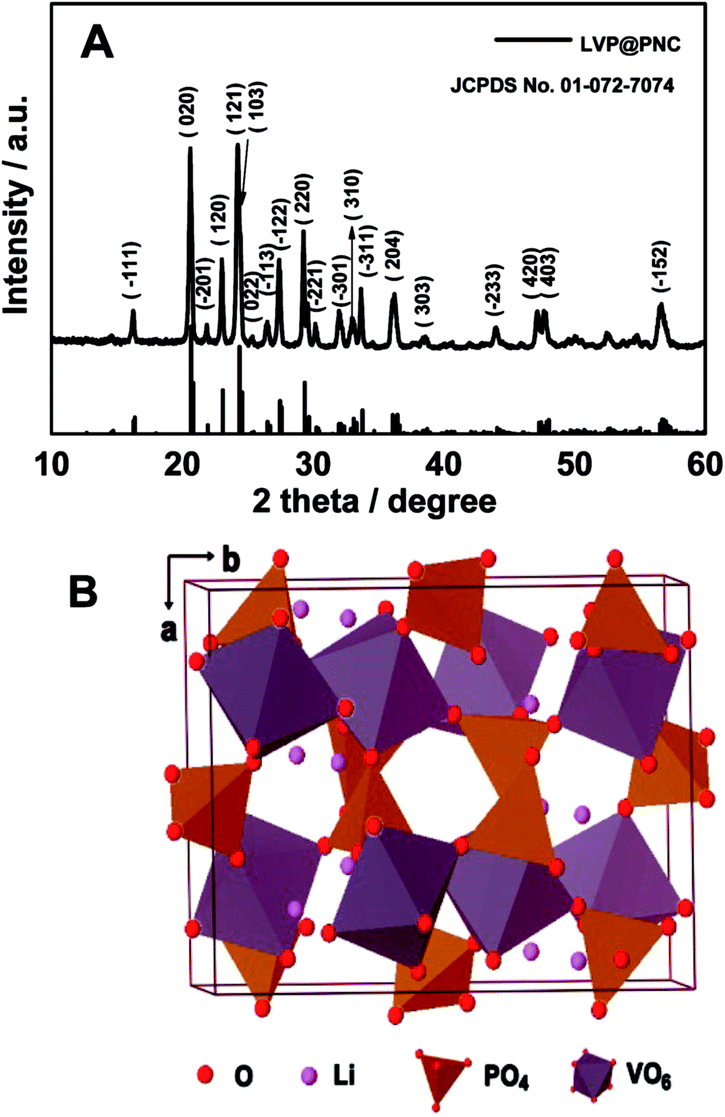 | ||
| Fig. 1 (A) XRD pattern of LVP@PNC, (B) crystal structure of monoclinic Li3V2(PO4)3. | ||
In order to observe the morphology of the as-synthesized products, SEM images were recorded (Fig. 2A and B). The as-synthesized product were mainly composed of micro sized particles and disordered large pores with a diameter around 300 nm. For further investing the texture of the material, TEM images were also performed (Fig. 2C). It is clear that there are a lot of pores with a diameter around 10 nm in the product. To obtain the specific surface areas of LVP@PNC, N2 adsorption/desorption isotherms were obtained as shown in Fig. S1.† The specific BET surface area is 8.6 m2 g−1. Furthermore, the surface area of the carbon layer is about 148 m2 g−1 by subtract the weight of LVP which surface area is negligible due to the large particle size, indicating the existence of porous carbon in the composite. As shown in the HRTEM image (Fig. 2D), the lattice fringes with a spacing of 0.54 nm can be attributed to the (−111) plane of monoclinic Li3V2(PO4)3.23,40
 | ||
| Fig. 2 (A) and (B) SEM images of LVP@PNC, (C) TEM image of LVP@PNC, (D) HRTEM image of LVP@PNC. | ||
In order to verify the presence of nitrogen in the porous carbon, X-ray photoelectron spectroscopy (XPS) was performed. As shown in Fig. 3A, the XPS spectra can be fitted into three peaks corresponding to pyridinic (398.7 eV), quaternary (401.0 eV), and pyrrolic (400.1 eV) nitrogen species, respectively.41 It suggests that nitrogen atoms have been successfully introduced into the carbon framework. The nitrogen atoms, doped in the carbon layer, could bring numerous edges or defects with high electronegativity.37,41,42
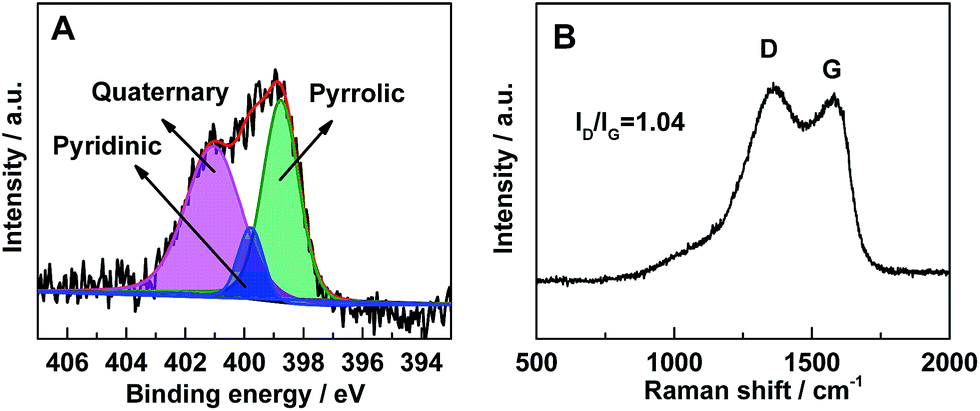 | ||
| Fig. 3 (A) N 1s XPS spectrum of LVP@PNC. In the XPS spectrum, the raw curve (black) is peak-fitted into three curves (indicated by pink, blue and green shaded areas) which related to three different types of N species (quaternary, pyridinic and pyrrolic), and the red curve is the sum of all three peak-fitted curves, (B) Raman spectrum of LVP@PNC. | ||
To further investigate the carbon species in the composites, Raman spectrum (Fig. 3B) was collected in the range of 500–2000 cm−1. The two distinct broad bands centered at ∼1360 and ∼1580 cm−1 are corresponding to the D-band (disorder-induced phonon mode) and G-band (graphite band), respectively.43–45 The absence of the LVP related vibrations may owe to the shielding effect of the surface carbon layer on the particles.46 The phenomenon also indicates that the LVP particles are embedded in the carbon framework closely. The large ratio of ID/IG also indicates the existence of amorphous carbon. Based on the ICP-OES analysis result, the LVP content is about 94.7 wt% in the composites, which means the residual carbon material is about 5.3 wt%. To exactly determine the amount of carbon in the composites, TG analysis was carried out (Fig. S2†). The composite is composed of about 5.8 wt% of carbon, which is in accordance with result of the ICP-OES analysis.
According to the previous reports, V5+ could be reduced to V3+ to form crystalline LVP by organic compounds pyrolyzed at high temperature in inert atmosphere.16,17 At the same time, residual organic compounds would form the carbon layer. In our case, it should be noted that dicyandiamide played an important role in the formation of porous carbon. For comparison, LVP coated with carbon (LVP@C) was prepared under the similar conditions without the addition of dicyandiamide. As shown in the XRD pattern and TEM image (Fig. S3A and B†), LVP@C could be obtained without obvious porous structure in the composites.
To investigate the effect of C3N4 during the preparation process, dicyandiamide has been calcined at 500 °C and 750 °C under N2 atmosphere, respectively. The XRD pattern (Fig. S4†) of the product by calcining dicyandiamide at 500 °C illustrate that the product mainly consists of g-C3N4.47 To determine the stability of the obtained g-C3N4, it was further calcined at 750 °C. As a result, no solid product was obtained, indicating the instability of the obtained g-C3N4 at this temperature.
To further investigate structure of the intermediate obtained at 500 °C, the IR spectrum and TEM image are obtained as shown in Fig. S5A and B.† The broad IR band ranging from 1200 to 1700 cm−1 is corresponded to the typical stretching vibration modes of heptazine units,48 indicating the existence of g-C3N4 in the intermediate. The bands at 505, 635 and 970 cm−1 can be assigned to the vibration of bonds between V3+ and O2− in the isolated VO6 octahedra. The bands at 573 and 1047 cm−1 suggest the presence of P–O bonds of PO4 tetrahedra. The others bands around 1100 cm−1 can be attributed to the stretching modes of terminal PO4 units. The bands at 950 and 758 cm−1 related to V5+ ions in isolated VO6 octahedra were not observed in the final product, indicating that the V5+ ions have been completely reduced to V3+. These bands appeared in the intermediate due to the reservation of V5+ species.49–51 As shown in TEM (Fig. S5B†), the intermediate exhibits a nonporous feature.
Based on above results, a mechanism was proposed for the formation of the porous composites (Scheme 1). During the calcination, citric acid would be pyrolyzed to form amorphous carbon and to reduce V5+ to form LVP. Simultaneously, g-C3N4 was formed in the amorphous carbon via the pyrolysis of dicyandiamide at lower temperature (left). Further increased the temperature to 750 °C, most of g-C3N4 would be decomposed, leaving plenty of pores and nitrogen atoms in the composites (right).52
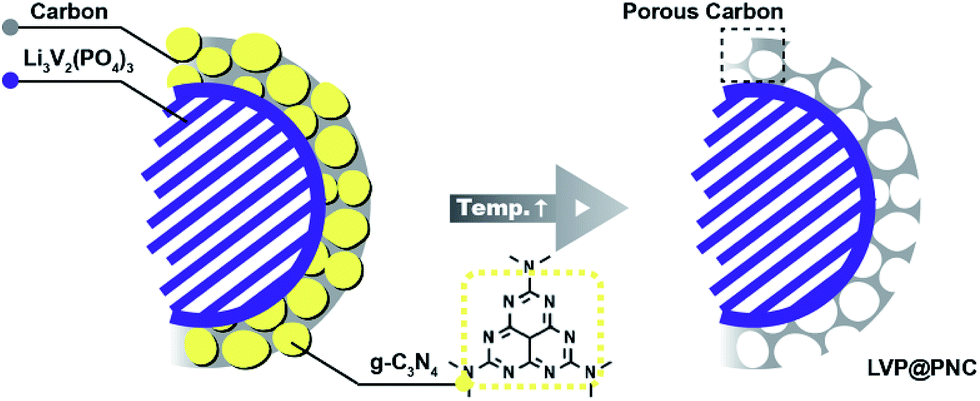 | ||
| Scheme 1 Schematic graph for the formation of LVP@PNC. | ||
In order to evaluate the electrochemical performance of LVP@PNC, electrochemical tests were carried out. The cyclic voltammetry (CV) curves for LVP@PNC were presented in Fig. 4A, which were recorded at a scan rate of 0.2 mV s−1 in the voltage range of 3.0 to 4.3 V (vs. Li/Li+) at room temperature. In the first charge process, the anodic peaks located at 3.62, 3.70 and 4.12 V are corresponded to the extraction of lithium ion. The first two anodic peaks are related to the first lithium ion extraction. While the second lithium ion extracts due to a single phase step corresponding to the last anodic peak. And the result is in accordance with the previous reports.50,53 In the first discharge process, the peaks located at 3.54, 3.62, 4.01 V are corresponding to the reversible lithiation of the LVP host coupling with the V4+/V3+ redox reaction.20 No obvious differences were found for these CV curves, illustrating the good stability of LVP@PNC.
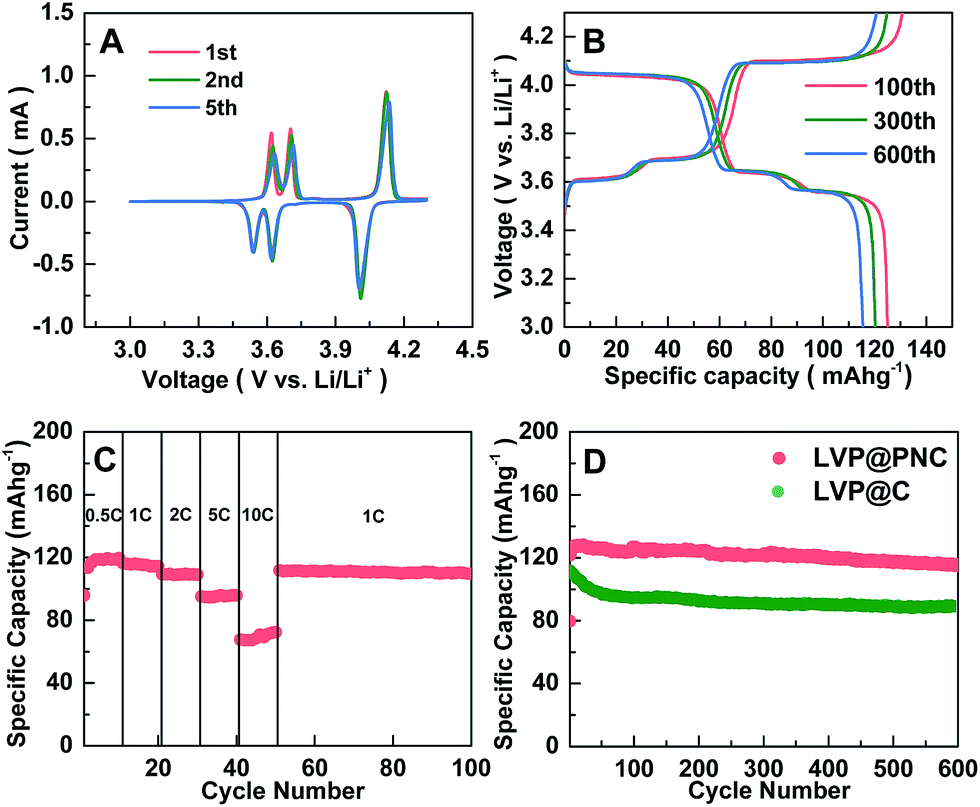 | ||
| Fig. 4 (A) CV curves of LVP@PNC at a scan rate of 0.2 mV s−1; (B) the specific charge–discharge capacities curves of LVP@PNC at 1 C; (C) the specific discharge capacities of LVP@PNC at different rates from 0.5 C to 10 C and (D) the cycling performance of LVP@PNC and LVP@C at the rate of 1 C for 600 cycles. | ||
The charge–discharge performances of LVP@PNC are presented in Fig. 4B. The three charge and discharge voltage plateaus in each cycle are related to the phase transition processes during the reversible lithium extraction and insertion of the LVP@PNC. It is in agreement with the CV curves presented in Fig. 4A. The tiny loss of specific discharge capacities and no obvious change in the charge–discharge curves also indicate the good cycling stability and low electrochemical polarization.
In Fig. 4C, the rate performance of LVP@PNC was tested at different charge/discharge rates from 0.5 C to 10 C. It delivers a relatively high reversible discharge capacity at different current rates (118.5, 115.1, 108.8, 95.0 and 72.4 mA h g−1 at 0.5, 2, 5 and 10 C, respectively). After the current rate goes back to 1 C, the reversible discharge capacity promptly returns to 111.6 mA h g−1. Even after 500 cycles, the reversible discharge capacity remains 100.5 mA h g−1 (Fig. S6†), indicating the good electrochemical reversibility of LVP@PNC.
Fig. 4D presents the long-term cycling performances of LVP@PNC at 1 C for 600 cycles. For comparison, LVP@C was also cycled under the same conditions. After the activation of the electrode for several cycles,54 a stable reversible discharge capacity of 114.7 mA h g−1 for LVP@PNC is obtained after 600 cycles at 1 C, whereas that of LVP@C is only 88.5 mA h g−1. To further evaluate the service life, the cycling performances of LVP@PNC and LVP@C are shown in Fig. S7 and S8.† The discharge capacities of LVP@PNC can reach 110.5, 90.5 and 79.8 mA h g−1 at 2, 5 and 10 C after 600 cycles, respectively, whereas those of LVP@C are 73.3, 51.3 and 50.3 mA h g−1. For comparison, some typical carbon coated Li3V2(PO4)3 cathode materials reported previously are summarized in Table S1.† Although some nanostructured LVP@C composites show high capacities and good cycling performances, the capacity and the long-term cycling performance (>600 cycles at high current rate of >5 C) of LVP@PNC are superior to those of carbon or porous carbon coated LVP with similar particle size.17,27,29,35,55–61 Both porous carbon layer and N-doped benefit the superior electrochemical performance of LVP@PNC.
Conclusions
In summary, LVP@PNC has been synthesized via a facile modified sol–gel method by using dicyandiamide as the necessary additive. The dicyandiamide-derived carbon nitride formed in situ plays dual roles. It can not only promote the formation of porous structure, but also act as nitrogen source for introducing nitrogen dopants in porous carbon. The as-obtained LVP@PNC cathode material exhibits high rate and good cycling performance for LIBs. We believed that this strategy can be used for the preparation of other porous carbon coating cathodes or anodes materials to enhance their performance.Acknowledgements
This work was supported by the NSFC (21371070, 21401066, 21401016).Notes and references
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- J. B. Goodenough and K. S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS PubMed.
- M. V. Reddy, G. V. Subba Rao and B. V. R. Chowdari, Chem. Rev., 2013, 113, 5364–5457 CrossRef CAS PubMed.
- M. S. Whittingham, Chem. Rev., 2004, 104, 4271–4301 CrossRef CAS.
- J. Dang, F. Xiang, N. Gu, R. Zhang, R. Mukherjee, I.-K. Oh, N. Koratkar and Z. Yang, J. Power Sources, 2013, 243, 33–39 CrossRef CAS PubMed.
- B. L. Ellis, K. T. Lee and L. F. Nazar, Chem. Mater., 2010, 22, 691–714 CrossRef CAS.
- W. Yuan, J. Yan, Z. Tang, O. Sha, J. Wang, W. Mao and L. Ma, J. Power Sources, 2012, 201, 301–306 CrossRef CAS PubMed.
- X. Du, W. He, X. Zhang, Y. Yue, H. Liu, X. Zhang, D. Min, X. Ge and Y. Du, J. Mater. Chem., 2012, 22, 5960–5969 RSC.
- A. K. Padhi, K. S. Nanjundaswamy and J. B. Goodenough, J. Electrochem. Soc., 1997, 144, 1188–1194 CrossRef CAS PubMed.
- K. Amine, H. Yasuda and M. Yamachi, Electrochem. Solid-State Lett., 2000, 3, 178–179 CrossRef CAS PubMed.
- L. Dimesso, D. Becker and C. Spanheimer, J. Solid State Electrochem., 2012, 16, 3791–3798 CrossRef CAS.
- J. Wolfenstine and J. Allen, J. Power Sources, 2004, 136, 150–153 CrossRef CAS PubMed.
- V. Aravindan, J. Gnanaraj, Y. Lee and S. Madhavi, J. Mater. Chem. A, 2013, 1, 3518–3539 CAS.
- S. Yang, P. Y. Zavalij and M. S. Whittingham, Electrochem. Commun., 2001, 3, 505–508 CrossRef CAS.
- H. Huang, S. C. Yin, T. Kerr, N. Taylor and L. F. Nazar, Adv. Mater., 2002, 14, 1525–1528 CrossRef CAS.
- X. Rui, Q. Yan, M. S. Kazacos and T. M. Lim, J. Power Sources, 2014, 258, 19–38 CrossRef CAS PubMed.
- Y. Luo, X. Xu, Y. Zhang, Y. Pi, Y. Zhao, X. Tian, Q. An, Q. Wei and L. Mai, Adv. Energy Mater., 2014, 4, 1–8 Search PubMed.
- J. Su, X. Wu, J. Lee, J. Kim and Y. Guo, J. Mater. Chem. A, 2013, 1, 2508–2514 CAS.
- X. H. Rui, D. H. Sim, K. M. Wong, J. X. Zhu, W. L. Liu, C. Xu, H. T. Tan, N. Xiao, H. H. Hng, T. M. Lim and Q. Y. Yan, J. Power Sources, 2012, 214, 171–177 CrossRef CAS PubMed.
- L. Mai, S. Li, Y. Dong, Y. Zhao, Y. Luo and H. Xu, Nanoscale, 2013, 5, 4864–4869 RSC.
- S. C. Yin, H. Grondey, P. Strobel, M. Anne and L. F. Nazar, J. Am. Chem. Soc., 2003, 125, 10402–10411 CrossRef CAS PubMed.
- M. M. Ren, Z. Zhou, Y. Z. Li, X. P. Gao and J. Yan, J. Power Sources, 2006, 162, 1357–1362 CrossRef CAS PubMed.
- C. Sun, S. Rajasekhara, Y. Dong and J. B. Goodenough, ACS Appl. Mater. Interfaces, 2011, 3, 3772–3776 CAS.
- L. Zhang, H. F. Xiang, Z. Li and H. H. Wang, J. Power Sources, 2012, 203, 121–125 CrossRef CAS PubMed.
- R. Zhang, Y. Zhang, K. Zhu, F. Du, Q. Fu, X. Yang, Y. Wang, X. Bie, G. Chen and Y. Wei, ACS Appl. Mater. Interfaces, 2014, 6, 12523–12530 CAS.
- M. S. Choi, H. S. Kim, Y. M. Lee and B. S. Jin, J. Mater. Chem. A, 2014, 2, 7873–7879 CAS.
- Q. Wei, Q. An, D. Chen, L. Mai, S. Chen, Y. Zhao, K. M. Hercule, L. Xu, A. M. Khan and Q. Zhang, Nano Lett., 2014, 14, 1042–1048 CrossRef CAS PubMed.
- H. Chen, Z. K. Wang, G. D. Li, F. F. Guo, M. H. Fan, X. Y. Wu and X. C. Cao, RSC Adv., 2015, 5, 31410–31414 RSC.
- C. Wang, H. Liu and W. Yang, J. Mater. Chem., 2012, 22, 5281–5285 RSC.
- K. Nathiya, D. Bhuvaneswari, Gangulibabu, D. Nirmala and N. Kalaiselvi, RSC Adv., 2012, 2, 6885–6889 RSC.
- X. L. Wu, L. Y. Jiang, F. F. Cao, Y. G. Guo and L. J. Wan, Adv. Mater., 2009, 21, 2710–2714 CrossRef CAS PubMed.
- X. Qin, H. Zhang, J. Wu, X. Chu, Y. B. He, C. Han, C. Miao, S. Wang, B. Li and F. Kang, Carbon, 2015, 87, 347–356 CrossRef CAS PubMed.
- H. Ni, J. Liu and L. Z. Fan, Nanoscale, 2013, 5, 2164–2168 RSC.
- H. J. Zhang, K. X. Wang, X. Y. Wu, Y. M. Jiang, Y. B. Zhai, C. Wang, X. Wei and J. S. Chen, Adv. Funct. Mater., 2014, 24, 3399–3404 CrossRef CAS PubMed.
- S. Wang, Z. Zhang, Z. Jiang, A. Deb, L. Yang and S. Hirano, J. Power Sources, 2014, 253, 294–299 CrossRef CAS PubMed.
- D. Li, M. Tian, R. Xie, Q. Li, X. Fan, L. Gou, P. Zhao, S. Ma, Y. Shi and H. T. H. Yong, Nanoscale, 2014, 6, 3302–3308 RSC.
- C. Wang, W. Shen and H. Liu, New J. Chem., 2014, 38, 430–436 RSC.
- C. Wang, Z. Guo, W. Shen, A. Zhang, Q. Xu, H. Liu and Y. Wang, J. Mater. Chem. A, 2015, 3, 6064–6072 CAS.
- J. Wang, C. Zhang and F. Kang, ACS Appl. Mater. Interfaces, 2015, 7, 9185–9194 CAS.
- L. Chen, C. Wang, H. Wang, E. Qiao, S. Wang, X. Jiang and G. Yang, Electrochim. Acta, 2014, 125, 338–346 CrossRef CAS PubMed.
- S. Maldonado, S. Morin and K. J. Stevenson, Carbon, 2006, 44, 1429–1437 CrossRef CAS PubMed.
- X. Zou, X. Huang, A. Goswami, R. Silva, B. R. Sathe, E. Mikmeková and T. Asefa, Angew. Chem., Int. Ed., 2014, 53, 4372–4376 CrossRef CAS PubMed.
- W. Duan, Z. Hu, K. Zhang, F. Cheng, Z. Tao and J. Chen, Nanoscale, 2013, 5, 6485–6490 RSC.
- Z. Zheng, Y. Wang, A. Zhang, T. Zhang, F. Cheng, Z. Tao and J. Chen, J. Power Sources, 2012, 198, 229–235 CrossRef CAS PubMed.
- L. Fei, W. Lu, L. Sun, J. Wang, J. Wei, H. L. W. Chana and Y. Wang, RSC Adv., 2013, 3, 1297–1301 RSC.
- C. M. Burba and R. Frech, Solid State Ionics, 2007, 177, 3445–3454 CrossRef CAS PubMed.
- S. Hu, L. Ma, H. Wang, L. Zhang, Y. Zhao and G. Wu, RSC Adv., 2015, 5, 31947–31953 RSC.
- X. Zou, J. Su, R. Silva, A. Goswami, B. R. Satheab and T. Asefa, Chem. Commun., 2013, 49, 7522–7524 RSC.
- Z. Chen, C. Dai, G. Wu, M. Nelson, X. Hu, R. Zhang, J. Liu and J. Xia, Electrochim. Acta, 2010, 55, 8595–8599 CrossRef CAS PubMed.
- R. von Hagen, A. Lepcha, X. Song, W. Tyrra and S. Mathur, Nano Energy, 2013, 2, 304–313 CrossRef CAS PubMed.
- L. Wang, X. Jiang, X. Li, X. Pi, Y. Ren and F. Wu, Electrochim. Acta, 2010, 55, 5057–5062 CrossRef CAS PubMed.
- R. L. Xie, Z. M. Zong, F. J. Liu, Y. G. Wang, H. L. Yan, Z. H. Wei, M. Mayyasa and X. Y. Wei, RSC Adv., 2015, 5, 45718–45724 RSC.
- B. Pei, Z. Jiang, W. Zhang, Z. Yang and A. Manthiram, J. Power Sources, 2013, 239, 475–482 CrossRef CAS PubMed.
- W. F. Mao, C. X. Ma, Y. Ma and Z. Y. Tang, Ionics, 2014, 20, 897–900 CrossRef CAS.
- P. Xiong, L. Zeng, H. Li, C. Zheng and M. Wei, RSC Adv., 2015, 5, 57127–57132 RSC.
- L. Fei, L. Sun, W. Lu, M. Guo, H. Huang, J. Wang, H. L. W. Chan, S. Fan and Y. Wang, Nanoscale, 2014, 6, 12426–12433 RSC.
- J. Xu, S. L. Chou, C. Zhou, Q. F. Gu, H. K. Liu and S. X. Dou, J. Power Sources, 2014, 246, 124–131 CrossRef CAS PubMed.
- J. Su, X. L. Wu, J. S. Lee, J. Kimb and Y. G. Guo, J. Mater. Chem. A, 2013, 1, 2508–2514 CAS.
- Y. Q. Qiao, X. L. Wang, Y. J. Mai, J. Y. Xiang, D. Zhang, C. D. Gu and J. P. Tu, J. Power Sources, 2011, 196, 8706–8709 CrossRef CAS PubMed.
- L. Wang, X. Zhou and Y. Guo, J. Power Sources, 2010, 195, 2844–2850 CrossRef CAS PubMed.
- X. Zhou, Y. Liu and Y. Guo, Solid State Commun., 2008, 146, 261–264 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra14943f |
| This journal is © The Royal Society of Chemistry 2015 |