
A family of ligand and anion dependent structurally diverse Cu(ii) Schiff-base complexes and their catalytic efficacy in an O-arylation reaction in ethanolic media†
Abstract
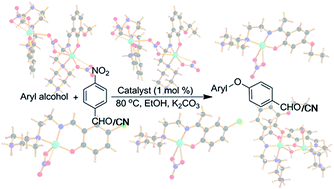
Two nitrato bridged dinuclear systems [Cu2(L1)2(NO3)3]NO3·H2O (1) and [Cu2(L2)2(NO3)3]NO3·MeOH (2), five monomeric complexes viz. [Cu(L3)(NO3)]NO3 (3), [Cu(L4)(NO3)]NO3 (4), [Cu(L5)(NO3)]NO3 (5), [Cu(L6)(NO3)NO3] (7), [Cu(L7)(NO3)]NO3 (8) and one hetero bi-bridged (phenoxido and water) dinuclear complex [Cu2(L2)2(H2O)2](ClO4)4·4H2O (6) have been synthesized and characterized using several physicochemical methods (L1 = 1-(N-3-methoxysalicylideneimino)-ethane-2-piperazine, L2 = 1-(N-3-ethoxysalicylideneimino)-ethane-2-piperazine, L3 = 1-(N-4′-ethoxy-α-methylasalicylideneimino)-ethane-2-piperazine, L4 = 1-(N-5′-chloro-α-methylasalicylideneimino)-ethane-2-piperazine, L5 = 1-(N-5-chlorosalicylideneimino)-ethane-2-piperazine, L6 = 1-(N-4-methoxysalicylideneimino)-ethane-2-piperazine and L7 = 1-(N-4′-methoxy-α-methylasalicylideneimino)-ethane-2-piperazine). X-ray structural analysis showed that complexes 1 and 2 are discrete dinuclear species where the pentacoordinated metal centers are bridged through a nitrate ion. In 3, 4, 5 and 8 the monomeric copper center displays a square pyramidal geometry with a weak axial Cu–O bond. In 7, the monomeric copper center shows a distorted octahedral geometry with two coordinated nitrate anions. However, in 6 the two copper centers coordinate in different manners (one is square-pyramidal and the other is distorted octahedral) and are bridged through a phenoxido group and a water molecule. All complexes efficiently catalyze the C–O coupling reaction under homogeneous conditions at 80 °C to afford unsymmetrical diaryl ethers using nitroarenes to act as an excellent electrophile. Notably, the reaction is carried out in ethanol media which facilitates the avoidance of toxic wastes. Structurally diverse copper(II) Schiff-base complexes have rarely been used systematically in catalytic C–O coupling reactions.
 Please wait while we load your content...
Please wait while we load your content...

