
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLanthanide complex-incorporated periodic mesoporous organosilica nanospheres with tunable photoluminescence†
Ning
Yuan
a,
Yucang
Liang
*a,
Egil Severin
Erichsen
b and
Reiner
Anwander
*a
aInstitut für Anorganische Chemie, Eberhard Karls Universität Tübingen, Auf der Morgenstelle 18, 72076 Tübingen, Germany. E-mail: yucang.liang@uni-tuebingen.de; reiner.anwander@uni-tuebingen.de
bLaboratory for Electron Microscopy, University of Bergen, Allégaten 41, 5007 Bergen, Norway
First published on 23rd September 2015
Abstract
A series of nanoscale periodic mesoporous organosilica spheres (PMOSs) have been synthesized via the co-condensation of N,N-bis(trimethoxysilylpropyl)-2,6-pyridine dicarboxamide (DPA-Si) and tetraethyl orthosilicate (TEOS) in the presence of the surfactant hexadecyltrimethylammonium bromide (CTAB) under alkaline conditions. All PMOSs were characterized by FT-IR and solid state 13C and 29Si MAS NMR spectroscopy, powder X-ray diffraction (PXRD) as well as nitrogen physisorption, confirming the successful incorporation of the bridging functional organic DPA moieties and the formation of ordered mesostructures. Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) revealed a spherical morphology (diameter ca. 70 nm) and ordered structures of the PMOSs. Incorporation of Eu(III) or Tb(III) ions into the PMOS frameworks by coordination to the DPA “ligands” generated lanthanide-hybrid PMOS with tunable photoluminescent properties that could make them promising candidates for applications in optical devices.
Introduction
Periodic mesoporous silicas (PMSs) have emerged as intriguing (host) materials due to their high specific surface area, uniform pore diameter, large pore volume, various topologies, and controlled morphologies.1–3 Geared to the seminal discovery of such purely siliceous PMSs, materials scientists and chemists alike have put tremendous efforts into developing respective materials carrying functional groups.4–6 As a result PMS post-treatment and co-condensation of tetraethyl orthosilicate (TEOS) with functional organosilane molecules as well as other approaches accomplished (metal)organogroup-functionalized inorganic–organic hybrid materials,5,7–11 which have been widely applied in heterogeneous catalysis,1,12 drug delivery,13,14 biomedical imaging,15,16 adsorption,17 and sensing.18–20 The synthesis methods, comprising simple doping and immobilization protocols, are categorized according to the interactions between the (metal)organic components and silica host.21 Hybrid materials fabricated by the former approach often suffer from leakage of the (metal)organic functional groups, due to their weak interactions with the host, such as electrostatic forces and hydrogen bonding.21,22 By contrast, the introduction of (metal)organic components via covalent attachment using grafting as well as co-condensation (one-pot synthesis) approaches successfully impeded any leaching issues. In the small mesopore regime, however, the “dangling” (protruding) functionalities often led to restricted pore access (pore blocking phenomena).7Deliberate choice of bridged organosilanes as silica source can subtly counteract such pore blocking and has established a new class of periodic mesoporous silica, so-called periodic mesoporous organosilicas (PMOs).23–25 Every silicon atom in PMO materials is connected to three oxygen atoms and one carbon atom of a bridging organic moiety. The organic functionality now acts as an integral part of the framework showing a regular and uniform distribution. Controlling the functional groups of such hybrid materials is beneficial to their applications in optical devices26 and catalytic performance.27 At present, a variety of organosilane derivatives, ranging from simple organic bridges to complicated functional groups, have been embedded within PMOs,27–29 displaying potential for applications in optics, adsorption and separation, and catalysis.30–33 Terminal or bridged pyridine derivative-based PMOs have been assessed only recently due to their strong coordination ability with metal ions and emerging fluorescent properties.34–39
Lanthanide complexes display excellent candidates for light-emitting applications because of their large Stokes shift, narrow-band emission, and high emission intensity.40–42 The most popular ions are trivalent europium and terbium due to their sizable energy gap, which makes them ideally suited for visible light emitting. Over the past decades, numerous Eu(III) and Tb(III) coordination compounds with strong luminescence have been designed and synthesized by employing ligands with aromatic chromophores to light up the central lanthanide ions through the well-known “antenna effect”.43,44 From the viewpoint of practical applications, luminescent lanthanide(III) complexes have been incorporated into tailor-made inorganic or organic solid hosts which can significantly improve their chemical and thermal stability.21,45–48 Particularly, sol–gel-derived mesoporous silica materials are popular candidates to be loaded with lanthanide complexes via simple doping or chemical immobilization. However, most reported lanthanide complex-incorporated hybrid silica materials prepared by grafting and co-condensation approaches were either amorphous or without any regular morphology and uniform particle size.
Recently, bridged 1,10-phenanthroline and 2,2-bipyridine-functionalized PMO materials were reported for chelation of lanthanide ions/complexes (Chart 1).38,39,49 The one-pot synthesis employing lanthanide complex and organosilane molecule in the presence of a structure-directing agent made all lanthanide complexes integrate into the silica framework or onto the surface of the pore walls. On the other hand, post-synthesis impregnation can be used to coordinate lanthanide ions to the functional organic moiety of the hybrid mesoporous materials affording stable lanthanide-modified luminescent materials that retain the ordered mesostructure and shape of the parent material. Hence, the synthesis of long-range-ordered mesoporous parent materials with uniform shape and size is crucial. Up to now, lanthanide complex-integrated hybrid PMOs with regular morphology and a particle size <100 nm are still very rare.21 Moreover, functional organosilica precursors capable of efficiently coordinating Ln(III) centers are not commercially available. Herein, we used N,N-bis(trimethoxysilylpropyl)-2,6-pyridine dicarboxamide (DPA-Si) and TEOS to fabricate functional nanosized PMOSs in the presence of hexadecyltrimethylammonium bromide (CTAB) under basic conditions. Coordination of lanthanide ions (Eu(III), Tb(III)) by donor atoms of the functional organic moiety of the hybrid mesoporous organosilica generated hybrid PMOSs with tunable photoluminescence (Scheme 1). The obtained hybrid materials were fully characterized and their optical properties were investigated in detail.
 | ||
| Chart 1 Representative bridged organosilane precursors for chelation of lanthanide ions/complexes reported in the literature.38,39,49 | ||
 | ||
| Scheme 1 Schematic diagram showing the preparation of lanthanide complex-incorporated luminescent PMOSs. A representative coordination of the Ln(III) centres is shown comprising DPA moieties, chloride ions and water as ligands. The grey, red, green and yellow porous spheres represent DPA-PMOS, Eu-DPA-PMOS, Tb-DPA-PMOS, and Eu/Tb-DPA-PMOS respectively. Ln = Eu or Tb, DPA = 2,6-pyridine dicarboxamide. | ||
Experimental section
Materials and measurements
All reagents, dipicolinic acid, (3-aminopropyl) trimethoxysilane, EuCl3·6H2O and TbCl3·6H2O from Alfa Aesar, CTAB, sodium hydroxide, concentrated HCl (37%), TEOS and ethanol from Sigma Aldrich, pyridine from TCI and thionyl chloride from Merck, were used as received. Diethyl ether was purified by using Grubbs columns (MBraun SPS, solvent purification system) before use.Powder X-ray diffraction (PXRD) patterns were obtained using a Bruker D8 ADVANCE instrument in the step/scan mode (step width = 0.00825; accumulation time = 2 s per step; range (2θ) = 0.50–10.00°) using monochromatic Cu-Kα radiation (λ = 1.5406 Å). 1H NMR spectra were recorded at 25 °C in CDCl3 solution on a Bruker-ADVANCE-DMX400 (5 mm BB, 1H: 400.13 MHz), and 1H chemical shifts were referenced to internal solvent resonances and reported in parts per million relative to TMS. 13C CP MAS NMR spectra were recorded at ambient temperature with a Bruker 200 MHz Ultrashield Plus instrument (4.7 T) equipped with CP magic-angle-spinning (MAS) hardware and using ZrO2 rotors with a diameter of 4 mm; 13C chemical shifts were referenced to glycine (13C: 50.288 ppm). 29Si MAS NMR spectra were obtained at ambient temperature on a Bruker ASX 300 instrument equipped with MAS hardware and using a ZrO2 rotor with an inside diameter of 7 mm. IR spectra were recorded on a NICOLET 6700 FTIR spectrometer using a DRIFT chamber with KBr/sample mixtures. Nitrogen physisorption was carried out on an ASAP 2020 volumetric adsorption apparatus (Micromeritics) at 77.4 K [am(N2, 77 K) = 0.162 nm2). All samples were degassed at an expected temperature under a vacuum <10−3 Torr for 6 h prior to measurement. The Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) methods were used to determine the specific surface areas and the pore size distributions of the samples, respectively. The excitation and emission spectra were recorded in Hellma® fluorescence quartz cuvette in ethanol at ambient temperature on a Varian Cary Eclipse Fluorescence Spectrophotometer. Scanning electron microscopy (SEM) was conducted on a JEOL JSM-5900LV microscope operated at an accelerating voltage of 15 kV. Transmission electron microscopy (TEM) observations were obtained using a JEOL JEM2010 operated at 160 kV. The lanthanide contents were measured via the inductively coupled plasma mass spectrometry (ICP-MS) method by WESSLING GmbH. Thermogravimetric analysis (TGA) measurements were performed on a Netzsch STA 449F3 instrument equipped with a quartz crucible at a heating rate of 2 K min−1 under Ar/O2/Ar atmosphere.
Synthesis of pyridine 2,6-dicarbonyl chloride
Pre-dried dipicolinic acid (5.014 g, 30 mmol) and thionyl chloride (50 ml, 688 mmol, 23 equiv.) were placed into an oven-dried round-bottom Schlenk flask and refluxed for 15 h under argon. After the reaction mixture had cooled to ambient temperature, unreacted thionyl chloride was distilled off under reduced pressure. The solid residue was further dried in a vacuum oven overnight to yield pyridine 2,6-dicarbonyl chloride as a white powder (5.6 g, 92%). 1H NMR (CDCl3, 400 MHz): 8.37 (d, 2H, Ar-H), 8.16 (t, 1H, Ar-H).Synthesis of N,N-bis(trimethoxysilylpropyl)-2,6-pyridine dicarboxamide (DPA-Si)
This organobridged silane precursor was synthesized according to a slightly modified literature procedure.50,51 Briefly, pyridine 2,6-dicarbonyl chloride (5.030 g, 24.6 mmol) was dissolved in dry diethyl ether (180 ml) in a round-bottom flask, and a mixture of (3-aminopropyl) trimethoxysilane (8.841 g, 49.3 mmol) and pyridine (4.291 g, 54.2 mmol) in dry diethyl ether (70 ml) was then added dropwise under argon. After having stirred at ambient temperature for 4 h, the reaction solution was filtered. The diethyl ether and pyridine were removed by rotary evaporation. N,N-Bis(trimethoxysilylpropyl)-2,6-pyridine dicarboxamide (DPA-Si) was obtained as a yellow oil in 65% yield (8 g, 16 mmol). 1H NMR (CDCl3, 400 MHz): 8.34 (d, 2H, Ar-H), 8.13 (bs, 2H, NH), 8.01 (t, 1H, Ar-H), 3.57 (s, 18H, CH3Si), 3.50 (t, 4H, CH2NH), 1.80 (m, 4H, CH2CH2CH2), 0.75 (t, 4H, CH2Si).Synthesis of PMOSs
PMOSs were synthesized by co-condensation of DPA-Si and TEOS using CTAB as a template and catalytic amounts of sodium hydroxide. In a typical procedure, CTAB (600 mg, 1.646 mmol) was stirred into a solution of water (288 g, 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) mol) and sodium hydroxide aqueous solution (2 M, 2.1 ml, 4.2 mmol). After having stirred at 80 °C for 30 min, a mixture of DPA-Si (66 mg, 0.1348 mmol) and TEOS (2.771 g, 13.30 mmol) in ethanol (1 ml) was added dropwise under vigorous stirring. The final suspension was continuously stirred at 80 °C for 2 h. A white powder was recovered by filtration (using several layers of filtration paper, 2–4 μm mesh), washed with water and ethanol twice, and dried in air. The surfactant was removed by solvent-extraction. The detailed procedure is as follows: as-synthesized material (ca. 1 g) was stirred into a solution of ethanol (200 ml) and concentrated hydrochloric acid (0.4 ml) at 65 °C for 4 h. This process was repeated twice to completely remove all surfactant. The obtained product was denoted as DPA-PMOS-1, for which the molar ratio of DPA-Si to TEOS was 1:99. By varying this ratio to either 2:98, 5:95 or 8:92, a series of DPA-PMOS materials denoted as DPA-PMOS-2, DPA-PMOS-3, and DPA-PMOS-4, were obtained, respectively. DRIFT (KBr, cm−1): 3340 (νO–H), 2980 and 2936 w (νas,C–H), 1667 (νC
mol) and sodium hydroxide aqueous solution (2 M, 2.1 ml, 4.2 mmol). After having stirred at 80 °C for 30 min, a mixture of DPA-Si (66 mg, 0.1348 mmol) and TEOS (2.771 g, 13.30 mmol) in ethanol (1 ml) was added dropwise under vigorous stirring. The final suspension was continuously stirred at 80 °C for 2 h. A white powder was recovered by filtration (using several layers of filtration paper, 2–4 μm mesh), washed with water and ethanol twice, and dried in air. The surfactant was removed by solvent-extraction. The detailed procedure is as follows: as-synthesized material (ca. 1 g) was stirred into a solution of ethanol (200 ml) and concentrated hydrochloric acid (0.4 ml) at 65 °C for 4 h. This process was repeated twice to completely remove all surfactant. The obtained product was denoted as DPA-PMOS-1, for which the molar ratio of DPA-Si to TEOS was 1:99. By varying this ratio to either 2:98, 5:95 or 8:92, a series of DPA-PMOS materials denoted as DPA-PMOS-2, DPA-PMOS-3, and DPA-PMOS-4, were obtained, respectively. DRIFT (KBr, cm−1): 3340 (νO–H), 2980 and 2936 w (νas,C–H), 1667 (νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1540 (δNH–νCN), 1070–1200 (νas,Si–O–Si), 949 (νas,Si–OH), 798 and 455 (νO–Si–O). Elemental analysis (wt%): C 8.20, H 2.46, N 1.11 for DPA-PMOS-1, C 8.94, H2.94, N 1.46 for DPA-PMOS-2, C 15.38, H 3.07, N 3.01 for DPA-PMOS-3, and C 17.39, H 4.00, N 4.02 for DPA-PMOS-4.
O), 1540 (δNH–νCN), 1070–1200 (νas,Si–O–Si), 949 (νas,Si–OH), 798 and 455 (νO–Si–O). Elemental analysis (wt%): C 8.20, H 2.46, N 1.11 for DPA-PMOS-1, C 8.94, H2.94, N 1.46 for DPA-PMOS-2, C 15.38, H 3.07, N 3.01 for DPA-PMOS-3, and C 17.39, H 4.00, N 4.02 for DPA-PMOS-4.
Synthesis of europium(III) or terbium(III)-incorporated periodic mesoporous organosilica nanospheres
In a typical procedure, to 25 ml of an ethanolic solution of EuCl3·6H2O (198 mg), a fine powder of DPA-PMOS-3 (250 mg) was added and stirred at 80 °C for 15 h. The product was collected by filtration, washed with an excess of ethanol, and dried in a vacuum oven to yield europium(III)-hybridized luminescent PMOS, Eu-DPA-PMOS-3. Employing TbCl3·6H2O instead of EuCl3·6H2O formed Tb-DPA-PMOS-3, whereas using an equimolar mixture of TbCl3·6H2O and EuCl3·6H2O gave Eu/Tb-DPA-PMOS-3. DRIFT (KBr, cm−1): 3332 (νO–H), 2980 and 2939 w (νas,C–H), 1653 (νCO), 1540 (δNH–νCN), 1070–1200 (νas,Si–O–Si), 953 (νas,Si–OH), 796 and 450 (νO–Si–O). Elemental analysis (wt%): C 15.66, H 2.94, N 2.99 for Eu-DPA-PMOS-3, C 15.71, H 3.17, N 2.99 for Tb-DPA-PMOS-3, C 14.50, H 2.71, N 2.91 for Eu/Tb-DPA-PMOS-3. ICP-MS (wt%): Eu 2.0 for Eu-DPA-PMOS-3, Tb 1.9 for Tb-DPA-PMOS-3 as well as Eu 0.89 and Tb 1.1 for Eu/Tb-DPA-PMOS-3.
Results and discussion
DPA-functionalized periodic mesoporous organosilica spheres (DPA-PMOSs)
The DPA-bridged bis(trimethoxysilane) (DPA-Si) under study was successfully synthesized according to slightly modified literature procedures and characterized by 1H NMR and FTIR spectroscopy. DPA moieties are known to efficiently coordinate rare-earth metal centres as proven by X-ray crystallography.52–56 A series of DPA-Si derivatives was previously used as ligands for lanthanide nitrate salts of Eu(III) and Gd(III) to prepare lanthanide-hybridized amorphous organosilica with luminescent properties.50 Moreover, hybrid materials obtained from co-condensation of TEOS with a DPA-Si derivative and subsequent treatment with europium(III) nitrate were examined for the fabrication of lanthanide-incorporated silica films.51 These hybrid materials did not possess any regular morphology but their long-range ordered structure was emphasized. In the present study, nanostructured DPA-functionalized PMOSs, denoted as DPA-PMOS-n, were prepared in a controlled manner using different ratios of TEOS and DPA-Si. The co-condensation was performed in the presence of CTAB as a structure-directing agent under basic conditions. The low angle PXRD patterns of the surfactant-free materials clearly showed a strong diffraction peak at a 2θ angle of about 2° indexed as (100) reflection and a poorly resolved broad peak at a 2θ angle of around 4° (Fig. 1) indicating the formation of a two-dimensionally ordered PMO of hexagonal symmetry.57,58 With increasing amounts of DPA-Si the intensity of the (100) reflection gradually decreased and the full width at half maximum slightly became broader, suggesting a lower long-range order of these materials.59 This can be attributed to the relatively large molecular size of the DPA-Si group, which affects the packing structure of the surfactant micelles in the reaction mixture via charge (mis)matching between surfactant CTAB and the functional inorganic silica framework. For comparison, Table 1 summarizes slight variations of the d-spacings and unit cell parameters a0 for all four materials.
 | ||
| Fig. 1 Low-angle PXRD patterns of (a) DPA-PMOS-1, (b) DPA-PMOS-2, (c) DPA-PMOS-3, and (d) DPA-PMOS-4. | ||
| Sample | d 100 a/nm | a 0 b/nm | a s,BET c/m2 g−1 | V p,ad d/cm3 g−1 | d p,ad e/nm | w p f/nm |
|---|---|---|---|---|---|---|
|
a Lattice spacings d100 = 1.5406/(2sinθ).
b Unit cell parameter a0 = 2d100/(3)1/2.
c Specific BET surface area.
d Single point adsorption total pore volume at relative pressure P/P0 = 0.99.
e Pore diameter according to the maximum of BJH pore size distribution from adsorption branch.
f Pore wall thickness wp = a0 − dp,ad.
|
||||||
| DPA-PMOS-1 | 4.32 | 4.98 | 977 | 1.16 | 2.4 | 2.58 |
| DPA-PMOS-2 | 4.23 | 4.89 | 942 | 1.37 | 2.3 | 2.59 |
| DPA-PMOS-3 | 4.21 | 4.87 | 921 | 1.32 | 2.2 | 2.67 |
| DPA-PMOS-4 | 4.18 | 4.83 | 727 | 0.99 | 2.1 | 2.78 |
| Eu-DPA-PMOS-3 | 3.93 | 4.54 | 870 | 1.21 | 2.2 | 2.34 |
| Tb-DPA-PMOS-3 | 3.96 | 4.57 | 859 | 1.29 | 2.2 | 2.37 |
| Eu/Tb-DPA-PMOS-3 | 3.96 | 4.57 | 861 | 1.22 | 2.2 | 2.37 |
In order to monitor the morphology and further confirm the ordered mesoporous structure SEM and TEM studies were performed on DPA-PMOS-x. Representative SEM and TEM images of materials DPA-PMOS-2 and DPA-PMOS-3 are shown in Fig. 2. For DPA-PMOS-2, uniform spheres with an average particle size of 67 nm were observed as well as chain-like aggregations of spheres (Fig. 2a). For DPA-PMOS-3 a similar particle morphology was found (Fig. 2c). The TEM image of DPA-PMOS-2 (Fig. 2b) clearly corroborates a long-range ordered mesoporous structure, while material DPA-PMOS-3 with an higher DPA-Si loading reveals a slightly decreased periodicity in the mesoporous structure (Fig. 2d).
 | ||
| Fig. 2 Representative SEM (a and c) and TEM (b and d) images for DPA-PMOS-2 (a and b) and DPA-PMOS-3 (c and d). All scale bars represent 100 nm. | ||
Moreover, incorporation of the functional DPA-Si moieties into the PMOS framework was confirmed by FTIR spectroscopy (Fig. 3). Characteristic stretching vibrations of the pure DPA-Si molecular precursor at 3340 cm−1 (νNH), 1680 cm−1 (νCO), 2975–2885 cm−1 (νCH,aliphatic) and at 3070 cm−1 (νCH,aromatic/pyridine) are known from literature.50,60 Hybrid materials DPA-PMOS-x (x = 1, 2, 3 and 4) showed very similar IR spectra (ESI, Fig. S1†) and that of DPA-PMOS-3 is representatively shown in Fig. 3b. The strong and broad band which appeared at around 1070–1200 cm−1 can be attributed to the asymmetric stretching vibrations of the Si–O–Si framework as well as the stretching vibrations of Si–C bonds originating from the hydrolysis and co-condensation of functional DPA-Si and TEOS. The bands at 455 and 798 cm−1 are assigned to the rocking and symmetric stretching vibrations of O–Si–O units, respectively, at 949 cm−1 to the asymmetric stretching vibration of the Si–OH group and in the range of around 3440–3740 cm−1 to characteristic O–H stretching vibrations of silanol and/or residual water molecules. While these findings confirm the formation of siloxane bonds and hydrolysis of TEOS and DPA-Si molecules, the characteristic absorption bands of CO (1667 cm−1) and δ(NH)–ν(CN) moieties (1540 cm−1)50 evidence intact DPA functionalities. Note that a red shift of the CO vibration (about 13 cm−1) and the band at 1590 cm−1 (pyridine ring) further prove the successful incorporation of the DPA-Si molecular unit. As expected, increasing amounts of the DPA-Si molecular precursor in the original reaction mixture, led to more intense absorption bands in the IR spectra of the hybrid materials (Fig. S1†).
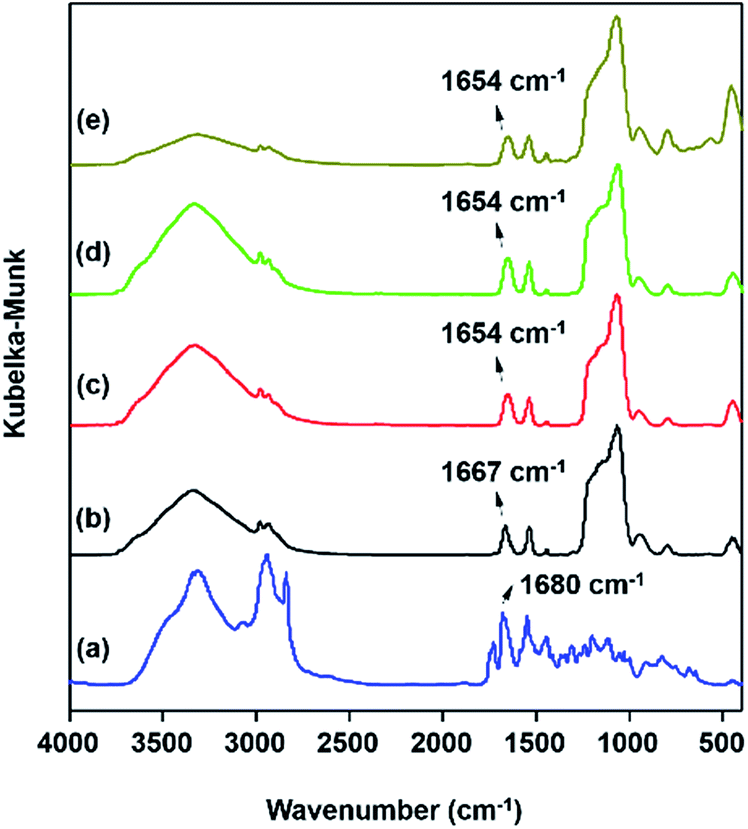 | ||
| Fig. 3 IR spectra of (a) precursor DPA-Si, (b) DPA-PMOS-3, (c) Eu-DPA-PMOS-3, (d) Tb-DPA-PMOS-3, and (e) Eu/Tb-DPA-PMOS-3. | ||
Furthermore, the functional DPA-Si group in obtained materials DPA-PMOS-x (x = 1, 2, 3, 4) can also be characterized by solid-state 13C and 29Si NMR spectroscopy. For example, the 13C CP MAS NMR spectrum of DPA-PMOS-3 shown in Fig. 4a clearly displays nine well-resolved signals revealing complete incorporation of DPA-Si molecular fragments without cleavage and hydrolysis of the amide groups.50 The chemical shifts can be assigned to the carbonyl group (δ = 166.4 ppm), the pyridine ring (δ = 150.6, 140.1, and 126.2 ppm), the propyl group (δ = 44.0, 24.7, and 11.7 ppm) as well as residual/unhydrolyzed ethoxyl groups of the TEOS precursor (δ = 59.6 and 18.1 ppm).
 | ||
| Fig. 4 (a) 13C and (b) 29Si CP MAS NMR spectra of DPA-PMOS-3. | ||
Solid-state 29Si MAS NMR spectroscopy is routinely used to confirm the covalent linkage of bridging organic groups within the hybrid materials and to assign the chemical environment of the silicon atoms. As shown in Fig. 4b for the 29Si CP MAS NMR spectrum, two different types of silicon species corresponding to Qn (Qn = Si(OSi)n(OH)4−n, n = 2, 3 and 4) and Tn (Tn = CSi(OSi)n(OH)3−n, n = 2 and 3) are observed, respectively. For the Qn-type silicon species, three signals at δ = −109.2, −100.2, and −91.2 ppm are assigned to the silicon resonances of Si(OSi)4 (Q4), HOSi(OSi)3 (Q3), and (HO)2Si(OSi)2 (Q2), respectively, showing a complete or partial condensation of pure silicon species of the mesoporous silica framework.61 The Tn-type silicon species observed at δ = −64.9 and −57.1 ppm can be assigned to the silicon resonances of completely condensed CSi(SiO)3 (T3) and of partially condensed CSi(SiO)2(OH) (T2), respectively, revealing the integration of the functional organic groups into the pore walls.25 When applying solid-state 29Si MAS NMR measurements using single-pulse excitation and high-power decoupling (Fig. S5†), the integral ratios of Tn to Qn for DPA-PMOS-2 and DPA-PMOS-3 could be determined as 0.048 and 0.092, respectively, which is very close to the molar ratios of initial DPA-Si precursor to TEOS, further confirming extensive hydrolysis of the DPA-Si precursor and a successful cross-linking with hydrolyzed silicon species from TEOS. This result can be used to quantitatively assess the degree of incorporation of the organic moiety.
To further characterize the pore structure of materials DPA-PMOS-x (x = 1, 2, 3, 4) nitrogen adsorption–desorption measurements were carried out. As can be seen in Fig. 5a, the nitrogen sorption isotherms of all materials are of type IV according to the IUPAC classification without clear hysteresis loops but with comparatively steep capillary condensation/evaporation steps at relative pressures in the range from 0.25 to 0.35,62 indicating a characteristic mesoporous structure with a narrow pore size distribution (Fig. 5b).63 Note that with increasing amounts of the DPA-Si precursor in the initial reaction mixture, the occurrence of the first capillary phenomenon of materials DPA-PMOS-x (x = 2, 3, 4) shifted slightly toward lower relative pressure, suggesting a reduction of the pore diameter. Moreover, for all materials, another condensation step was observed at high relative pressures (P/P0 > 0.90), indicating high textural porosity or the existence of interparticle porosity.64–67 Correspondingly, increasing DPA-Si contents considerably changed the pore parameters, such as the specific BET surface area (977 to 921 and finally to 727 m2 g−1), pore volume (1.37 to 0.99 cm3 g−1) and pore diameter (2.4 nm to 2.1 nm), as well as the pore wall thickness (2.6 to 2.8 nm) (Table 1). These findings further verified the successful integration of the functional organic moieties. Note that materials DPA-PMOS-2 and DPA-PMOS-3, which were obtained by DPA-Si precursor loadings of 2 and 5%, respectively, did not show any significant changes in structure and pore characteristics, although their chemical composition as confirmed by CHN elemental analysis is different. This suggests that at a certain level of bridging organic groups homogeneously integrated into the pore walls the topology and pore characteristics change only marginally. Because high surface areas and large pore volumes as well as high loadings of functional groups are beneficial for applications of materials in biomedicine, adsorption and separation, the material DPA-PMOS-3 was selected as a candidate for further coordination to lanthanide ions (Eu(III) and Tb(III)) to target photoluminescent PMOS materials.
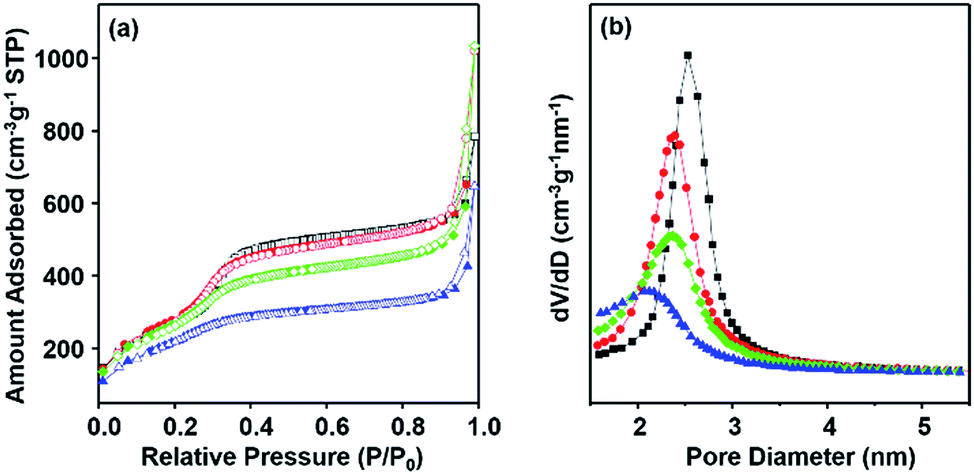 | ||
| Fig. 5 (a) Nitrogen adsorption–desorption isotherms and (b) the BJH pore size distributions of DPA-PMOS-1 (black squares), DPA-PMOS-2 (red circles), DPA-PMOS-3 (green diamonds), and DPA-PMOS-4 (blue triangles). | ||
Lanthanide-functionalized photoluminescent materials (Ln-DPA-PMOS-3, Ln = Eu, Tb, or Eu/Tb)
Lanthanide(III) ions were introduced into material DPA-PMOS-3 employing LnCl3·6H2O (Ln = Eu, Tb, or Eu/Tb) in ethanolic solution. Ln(III) coordination to nitrogen(pyridine) and oxygen(dicarboxamide) atoms of the covalently incorporated DPA-Si moiety was anticipated to generate photoluminescent hybrid materials Ln-DPA-PMOS-3 (Ln = Eu, Tb, Eu/Tb). Any free lanthanide ions were washed out with excessive amounts of solvent. The PXRD patterns of materials Ln-DPA-PMOS-3 (Ln = Eu, Tb, Eu/Tb) (Fig. 6) clearly display the preservation of the distinct diffraction signals belonging to the 2D hexagonal structure of the parent material DPA-PMOS-3. Although the ordering of the pore structure was not affected, the intensity of the reflection peaks markedly decreased due to loading and coordination of the lanthanide ions (ca. 2% from ICP analysis).68 | ||
| Fig. 6 Low angle PXRD patterns of (a) parent material DPA-PMOS-3, lanthanide-coordinated hybrid materials (b) Eu-DPA-PMOS-3, (c) Tb-DPA-PMOS-3, and (d) Eu/Tb-DPA-PMOS-3. | ||
As shown in Fig. 2c–e, the IR spectra of Eu-DPA-PMOS-3, Tb-DPA-PMOS-3, and Eu/Tb-DPA-PMOS-3 exhibit characteristic bands of the functional DPA moiety at 1654 (νCO) and 1540 cm−1 (δNH–νCN). A red-shift of the carbonyl stretching vibration (νCO) confirmed the coordination of the lanthanide ions.50
The nitrogen adsorption–desorption isotherms of Ln-DPA-PMOS-3 (Ln = Eu, Tb, Eu/Tb) are quite similar to that of the parent material DPA-PMOS-3, corroborating that Eu or Tb-incorporation did not markedly affect the regular mesoporous structure (Fig. 7). However, the corresponding specific BET surface area and pore volume decreased to 94.4 and 91.7% of the parent material for Eu-DPA-PMOS-3, 93.3 and 97.7% of the parent material for Tb-DPA-PMOS-3, and 93.5 and 92.4% of the parent material for Eu/Tb-DPA-PMOS-3, respectively (Table 1). Note that the pore diameter and pore wall thickness did not show any marked alterations, evidencing the coordination of the lanthanide ion to the DPA donor atoms and the absence of any aggregation of free lanthanide salt inside the pore channels. This is also evidenced by relatively narrow BJH pore size distributions (Fig. 7b).
 | ||
| Fig. 7 (a) Nitrogen adsorption–desorption isotherms and (b) the BJH pore size distribution of DPA-PMOS-3 (black squares), Eu-DPA-PMOS-3 (red circles), Tb-DPA-PMOS-3 (green diamonds), and Eu/Tb-DPA-PMOS-3 (yellow triangles). | ||
The photoluminescent properties of the lanthanide-coordinated hybrid materials Ln-DPA-PMOS-3 (Ln = Eu, Tb, Eu/Tb) were investigated at ambient temperature. The excitation and emission spectra are presented in Fig. S2† and 8, respectively. For Eu-DPA-PMOS-3, the excitation spectrum was obtained by monitoring the emission wavelength of Eu(III) ions at 615 nm. The broad and strong band ranging from 250 to 400 nm is assigned to the electron transitions from ground state to excited state (π–π*) of the incorporated organic DPA moiety.50 Due to the relatively weak intensity of the f–f transitions of Eu(III) ions bands in the longer wavelength region cannot be observed.69 Upon DPA-mediated excitation of the europium(III) hybrid material at 292 nm, the emission spectrum displayed the characteristic bands of Eu(III) ions centered at 590, 615, 651 and 700 nm, corresponding to 5D0 → 7Fi (i = 1, 2, 3, 4) intra-4f transitions.70 The dominant band located around 615 nm can be assigned to the electronic dipole transition 5D0 → 7F2 (hypersensitive transition), indicative of an asymmetric coordination environment of the Eu(III) ions and relatively pure red-color emission as well. The second strongest band at 590 nm with orange emission corresponds to the magnetic dipole transition 5D0 → 7F1.71 The ratio of intensity of 5D0 → 7F2 to 5D0 → 7F1 is practically used to assess the symmetry around Eu(III) ions. For Eu-DPA-PMOS-3, a ratio of 3.07 indicates a low-symmetry environment of the Eu(III) ions.72
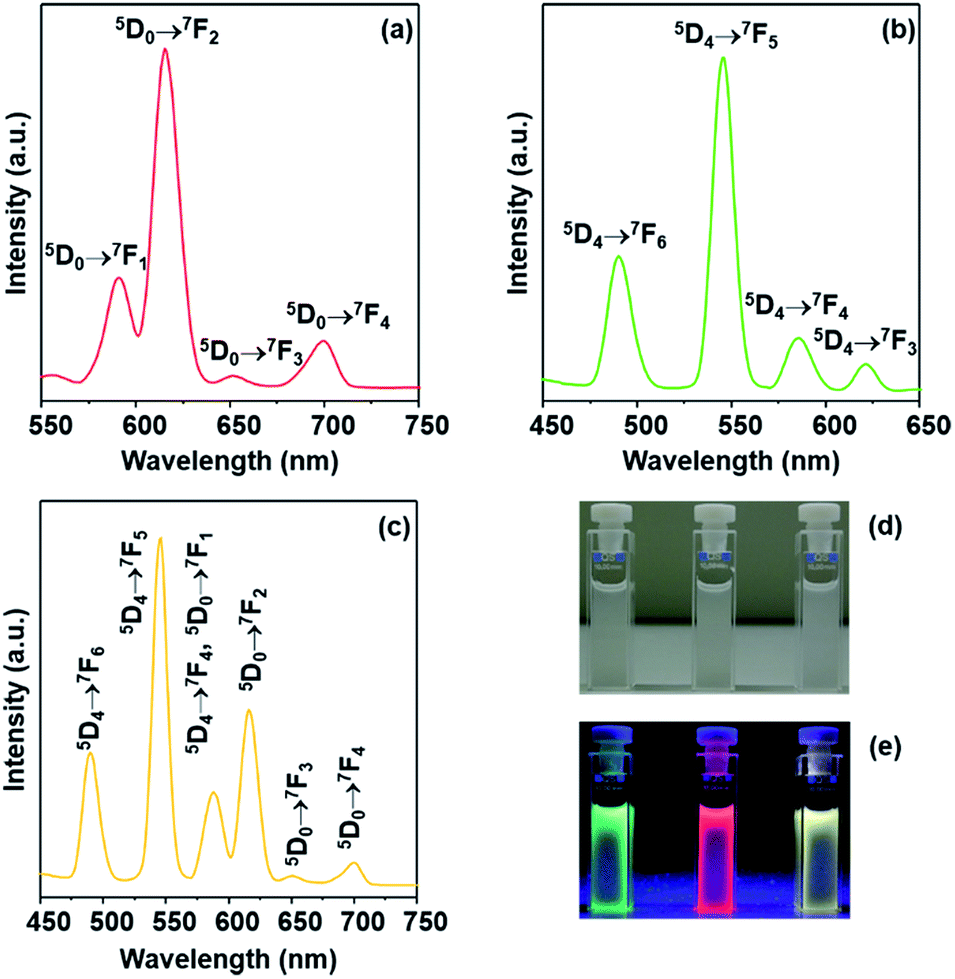 | ||
| Fig. 8 Emission spectra of (a) Eu-DPA-PMOS-3, (b) Tb-DPA-PMOS-3, and (c) Eu/Tb-DPA-PMOS-3 at ambient temperature. Images of Eu-DPA-PMOS-3 (middle), Tb-DPA-PMOS-3 (left), and Eu/Tb-DPA-PMOS-3 (right) under (d) sunlight lamp and (e) UV irradiation. | ||
For material Tb-DPA-PMOS-3, the excitation spectrum shown in Fig. S2b† was obtained by monitoring the 5D4 → 7F5 emission of Tb(III) ions at 545 nm. It is similar to that of the europium hybrid counterpart and displays a broad band peaked at 293 nm. The corresponding emission spectrum shown in Fig. 8b clearly display four major bands centered at 490, 545, 586 and 621 nm, which can be assigned to 5D4 → 7Fj (j = 6, 5, 4, 3) intra-4f transitions, characteristic emissions of Tb(III) ions.73 In addition, the dominant band at 545 nm corresponds to the magnetic dipole transition 5D4 → 7F5, indicative of typical green-color light emitting.
Coordinative bonding between organic ligands and a mixture of different lanthanide ions has been proposed as a strategy to modulate the emission.74–76Fig. 8c displays the emission spectrum of Eu/Tb-DPA-PMOS-3 upon excitation at 290 nm. Both the emission bands originating from Eu(III) and Tb(III) complexes appeared, with the two notable emission bands located at 545 nm and 615 nm corresponding to the 5D4 → 7F5 transition of the Tb(III) ions and 5D0 → 7F2 transition of the Eu(III) ions, respectively. Other emission bands could be assigned to respective intra-4f transitions of Tb(III) or Eu(III) ions. Moreover, upon UV irradiation (wavelength of 254 nm) of lanthanide-incorporated hybrid mesoporous organosilicas Eu-DPA-PMOS-3, Tb-DPA-PMOS-3, and Eu/Tb-DPA-PMOS-3 displayed brilliant red, green, and yellow emission, respectively, at ambient temperature (Fig. 8e). The corresponding UV-visible spectra are shown in Fig. S3.†
Finally, the thermal stability of the obtained materials Ln-DPA-PMOS-3 (Ln = Eu, Tb, Eu/Tb) was investigated by thermogravimetric analysis (TGA). As shown in Fig. 9, both the parent material DPA-PMOS-3 and the europium ion-incorporated product Eu-DPA-PMOS-3 display two stages of weight loss during temperature increase from 30 to 1000 °C. For DPA-PMOS-3, the first stage (ca. 6%), occurring below 150 °C, can be assigned to the evaporation of physisorbed water molecules. The subsequent mass-loss of ca. 22% between 330 and 600 °C is due to the decomposition of the organic components in this material. This weight loss is in accordance with the calculated data (25.9%) assuming that all the DPA-Si precursor and TEOS were hydrolyzed and co-condensed with each other. In comparison, for Eu-DPA-PMOS-3, the initial weight-loss of ca. 8% corresponds to the displacement of physisorbed and chemically coordinated water molecules. The second stage of weight loss (ca. 22%) starting from 360 °C is attributed to the decomposition of the incorporated DPA ligand and europium complex. The residual weight of about 70% can be ascribed to the Si–O silica network. Similar thermogravimetric behaviour was also observed for Tb-DPA-PMOS-3 and Eu/Tb-DPA-PMOS-3 (Fig. S4†). It is worth noting that the onset of decomposition of the organic components for Ln-DPA-PMOS-3 (360 °C) is at higher temperature than that of their parent material DPA-PMOS-3 (330 °C), which means that the introduction of lanthanide ions can improve the thermal stability of such PMOS materials.
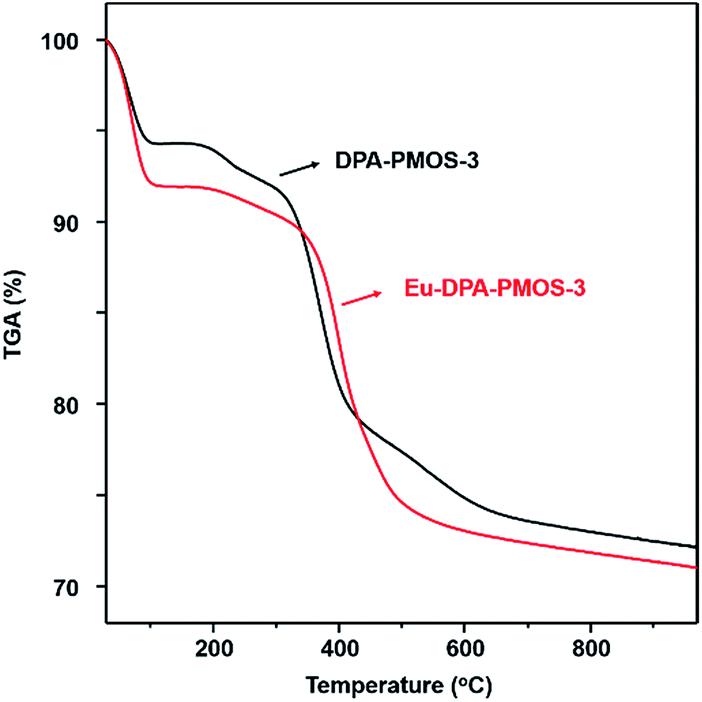 | ||
| Fig. 9 Thermogravimetric analysis (TGA) curves of DPA-PMOS-3 (black line) and Eu-DPA-PMOS-3 (red line). | ||
Conclusions
Functional DPA-Si (N,N-bis(trimethoxysilylpropyl)-2,6-pyridine dicarboxamide) can be successfully applied as covalent cross-linker in the co-condensation with TEOS to fabricate mesoporous silica frameworks of regular spherical morphology with characteristic hexagonal mesopore arrays. The DPA acid-amide bonds stayed intact during the sol–gel synthesis and post-treatments as evidenced by MAS NMR and FTIR spectroscopy. Optimal DPA loadings were obtained for initial DPA-Si/TEOS ratios of 2:98 and 5:95, as revealed by PXRD and nitrogen physisorption analyses. Coordination of lanthanide ions with the donor atoms of the DPA moieties afforded photoluminescent hybrid materials Ln-DPA-PMOS (Ln = Eu, Tb, Eu/Tb), exhibiting the characteristic emissions of the corresponding lanthanide(III) ions. The emission colours could be modulated by varying the employed lanthanide ions or by using a mixture of lanthanide ions. Given the small nanoscale spherical shape (diameter ca. 70 nm), high surface area, large pore volume as well as functional group loading (=photoluminescent properties), these nanoscale mesoporous organosilica spheres can be considered promising candidates for optical applications.
Acknowledgements
Ning Yuan acknowledges financial support from the China Scholarship Council (CSC). We are grateful to the University of Tübingen for funding within the program EXPAND, Dr Markus Ströbele for TGA measurements and Elke Nadler for SEM measurements.Notes and references
- Y. Liang and R. Anwander, Dalton Trans., 2013, 42, 12521 RSC.
- C. T. Kresge and W. J. Roth, Chem. Soc. Rev., 2013, 42, 3663 RSC.
- I. I. Slowing, J. L. Vivero-Escoto, B. G. Trewyn and V. S.-Y. Lin, J. Mater. Chem., 2010, 20, 7924 RSC.
- R. Anwander, Chem. Mater., 2001, 13, 4419 CrossRef CAS.
- A. Sayari and S. Hamoudi, Chem. Mater., 2001, 13, 3151 CrossRef CAS.
- P. Kumar and V. V. Guliants, Microporous Mesoporous Mater., 2010, 132, 1 CrossRef CAS.
- F. Hoffmann, M. Cornelius, J. Morell and M. Fröba, Angew. Chem., Int. Ed., 2006, 45, 3216 CrossRef CAS PubMed.
- N. Linares, E. Serrano, M. Rico, A. M. Balu, E. Losada, R. Luque and J. García-Martínez, Chem. Commun., 2011, 47, 9024 RSC.
- D. Brühwiler, Nanoscale, 2010, 2, 887 RSC.
- A. Mehdi, C. Reye and R. Corriu, Chem. Soc. Rev., 2011, 40, 563 RSC.
- A. Stein, B. J. Melde and R. C. Schroden, Adv. Mater., 2000, 12, 1403 CrossRef CAS.
- E. Le Roux, Y. Liang, M. P. Storz and R. Anwander, J. Am. Chem. Soc., 2010, 132, 16368 CrossRef CAS PubMed.
- C. Coll, A. Bernardos, R. Martínez-Máñez and F. Sancenón, Acc. Chem. Res., 2013, 46, 339 CrossRef CAS PubMed.
- B. G. Trewyn, I. I. Slowing, S. Giri, H.-T. Chen and V. S.-Y. Lin, Acc. Chem. Res., 2007, 40, 846 CrossRef CAS PubMed.
- J. L. Vivero-Escoto, R. C. Huxford-Phillips and W. Lin, Chem. Soc. Rev., 2012, 41, 2673 RSC.
- J. Wang, W. Guo, J.-H. Bae, S.-H. Kim, J. Song and C.-S. Ha, J. Mater. Chem., 2012, 22, 24681 RSC.
- Z.-A. Qiao, Q. Huo, M. Chi, G. M. Veith, A. J. Binder and S. Dai, Adv. Mater., 2012, 24, 6017 CrossRef CAS PubMed.
- B. J. Melde, B. J. Johnson and P. T. Charles, Sensors, 2008, 8, 5202 CrossRef CAS.
- B. J. Melde and B. J. Johnson, Anal. Bioanal. Chem., 2010, 398, 1565 CrossRef CAS PubMed.
- K. Hayashi, M. Nakamura, H. Miki, S. Ozaki, M. Abe, T. Matsumoto and K. Ishimura, Adv. Funct. Mater., 2012, 22, 3539 CrossRef CAS.
- J. Feng and H. Zhang, Chem. Soc. Rev., 2013, 42, 387 RSC.
- R. P. Bagwe, C. Yang, L. R. Hilliard and W. Tan, Langmuir, 2004, 20, 8336 CrossRef CAS PubMed.
- T. Asefa, M. MacLachlan, N. Coombs and G. Ozin, Nature, 1999, 402, 867 CAS.
- B. J. Melde, B. T. Holland, C. F. Blanford and A. Stein, Chem. Mater., 1999, 11, 3302 CrossRef CAS.
- S. Inagaki, S. Guan, Y. Fukushima, T. Ohsuna and O. Terasaki, J. Am. Chem. Soc., 1999, 121, 9611 CrossRef CAS.
- Y.-J. Li, L. Wang and B. Yan, J. Mater. Chem., 2011, 21, 1130 RSC.
- P. Van der Voort, D. Esquivel, E. De Canck, F. Goethals, I. Van Driessche and F. J. Romero-Salguero, Chem. Soc. Rev., 2013, 42, 3913 RSC.
- N. Mizoshita, T. Tani and S. Inagaki, Chem. Soc. Rev., 2011, 40, 789 RSC.
- B. Hatton, K. Landskron, W. Whitnall, D. Perovic and G. A. Ozin, Acc. Chem. Res., 2005, 38, 305 CrossRef CAS PubMed.
- E.-Y. Jeong and S.-E. Park, Res. Chem. Intermed., 2011, 38, 1237 CrossRef.
- F. Hoffmann and M. Fröba, Chem. Soc. Rev., 2011, 40, 608 RSC.
- W. Wang, J. E. Lofgreen and G. A. Ozin, Small, 2010, 6, 2634 CrossRef CAS PubMed.
- Q. Yang, J. Liu, L. Zhang and C. Li, J. Mater. Chem., 2009, 19, 1945 RSC.
- M. Waki, N. Mizoshita, T. Ohsuna, T. Tani and S. Inagaki, Chem. Commun., 2010, 46, 8163 RSC.
- A. Liu, S. Han, H. Che and L. Hua, Langmuir, 2010, 26, 3555 CrossRef CAS PubMed.
- M. S. Moorthy, M.-J. Kim, J.-H. Bae, S. S. Park, N. Saravanan, S.-H. Kim and C.-S. Ha, Eur. J. Inorg. Chem., 2013, 3028 CrossRef CAS.
- E. J. Cho, J. K. Kang and J. H. Jung, Mater. Lett., 2007, 61, 5157 CrossRef CAS.
- L.-N. Sun, H.-J. Zhang, C.-Y. Peng, J.-B. Yu, Q.-G. Meng, L.-S. Fu, F.-Y. Liu and X.-M. Guo, J. Phys. Chem. B, 2006, 110, 7249 CrossRef CAS PubMed.
- L. Sun, W. Mai, S. Dang, Y. Qiu, W. Deng, L. Shi, W. Yan and H. Zhang, J. Mater. Chem., 2012, 22, 5121 RSC.
- S. V. Eliseeva and J.-C. G. Bünzli, Chem. Soc. Rev., 2010, 39, 189 RSC.
- L. Armelao, S. Quici, F. Barigelletti, G. Accorsi, G. Bottaro, M. Cavazzini and E. Tondello, Coord. Chem. Rev., 2010, 254, 487 CrossRef CAS.
- L. D. Carlos, R. A. S. Ferreira, V. de Zea Bermudez and S. J. L. Ribeiro, Adv. Mater., 2009, 21, 509 CrossRef CAS PubMed.
- Y. Ma and Y. Wang, Coord. Chem. Rev., 2010, 254, 972 CrossRef CAS.
- J.-C. G. Bünzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048 RSC.
- L. D. Carlos, R. A. S. Ferreira, V. de Zea Bermudez, B. Julián-López and P. Escribano, Chem. Soc. Rev., 2011, 40, 536 RSC.
- K. Binnemans, Chem. Rev., 2009, 109, 4283 CrossRef CAS PubMed.
- N. Lin, H. Li, Y. Wang, Y. Feng, D. Qin, Q. Gan and S. Chen, Eur. J. Inorg. Chem., 2008, 4781 CrossRef CAS.
- H. Li, N. Lin, Y. Wang, Y. Feng, Q. Gan, H. Zhang, Q. Dong and Y. Chen, Eur. J. Inorg. Chem., 2009, 519 CrossRef CAS.
- X. Guo, X. Wang, H. Zhang, L. Fu, H. Guo, J. Yu, L. D. Carlos and K. Yang, Microporous Mesoporous Mater., 2008, 116, 28 CrossRef CAS.
- A.-C. Franville, D. Zambon, R. Mahiou and Y. Troin, Chem. Mater., 2000, 12, 428 CrossRef CAS.
- B. C. Barja, S. E. Bari, M. C. Marchi, F. L. Iglesias and M. Bernardi, Sens. Actuators, B, 2011, 158, 214 CrossRef CAS.
- B. E. Aroussi, S. Zebret, C. Besnard, P. Perrottet and J. Hamacek, J. Am. Chem. Soc., 2011, 133, 10764 CrossRef PubMed.
- J. P. Leonard, P. Jensen, T. McCabe, J. E. O'Brien, R. D. Peacock, P. E. Kruger and T. Gunnlaugsson, J. Am. Chem. Soc., 2007, 129, 10986 CrossRef CAS PubMed.
- K. T. Hua, J. Xu, E. E. Quiroz, S. Lopez, A. J. Ingram, V. A. Johnson, A. R. Tisch, A. de Bettencourt-Dias, D. A. Straus and G. Muller, Inorg. Chem., 2012, 51, 647 CrossRef CAS PubMed.
- O. Kotova, J. A. Kitchen, C. Lincheneau, R. D. Peacock and T. Gunnlaugsson, Chem.–Eur. J., 2013, 19, 16181 CrossRef CAS PubMed.
- A.-S. Chauvin, J.-C. G. Bünzli, F. Bochud, R. Scopelliti and P. Froidevaux, Chem.–Eur. J., 2006, 12, 6852 CrossRef CAS PubMed.
- P. Mohanty, Y. Fei and K. Landskron, J. Am. Chem. Soc., 2009, 131, 9638 CrossRef CAS PubMed.
- J. Huang, F. Zhu, W. He, F. Zhang, W. Wang and H. Li, J. Am. Chem. Soc., 2010, 132, 1492 CrossRef CAS PubMed.
- C. D. Nunes, P. D. Vaz, P. Brandão, J. Rocha, P. Ferreira, N. Bion and M. J. Calhorda, Microporous Mesoporous Mater., 2006, 95, 104 CrossRef CAS.
- A.-C. Franville, R. Mahiou, D. Zambon and J.-C. Cousseins, Solid State Sci., 2001, 3, 211 CrossRef CAS.
- E.-Y. Jeong, A. Burri, S.-Y. Lee and S.-E. Park, J. Mater. Chem., 2010, 20, 10869 RSC.
- D. H. Everett, Pure Appl. Chem., 1972, 31, 579 CrossRef.
- Z. Zhou, A. W. Franz, M. Hartmann, A. Seifert, T. J. J. Müller and W. R. Thiel, Chem. Mater., 2008, 20, 4986 CrossRef CAS.
- Y. Han and J. Y. Ying, Angew. Chem., Int. Ed., 2004, 44, 288 CrossRef PubMed.
- A. B. Fuertes, J. Mater. Chem., 2003, 13, 3085 RSC.
- S. H. Joo, R. Ryoo, M. Kruk and M. Jaroniec, J. Phys. Chem. B, 2002, 106, 4640 CrossRef CAS.
- E. Prouzet, F. Cot, G. Nabias, A. Larbot, P. Kooyman and T. J. Pinnavaia, Chem. Mater., 1999, 11, 1498 CrossRef CAS.
- X. Guo, H. Guo, L. Fu, H. Zhang, R. Deng, L. Sun, J. Feng and S. Dang, Microporous Mesoporous Mater., 2009, 119, 252 CrossRef CAS.
- S. Huang, Z. Cheng, P. Ma, X. Kang, Y. Dai and J. Lin, Dalton Trans., 2013, 42, 6523 RSC.
- Q. Xu, L. Li, X. Liu and R. Xu, Chem. Mater., 2002, 14, 549 CrossRef CAS.
- P. Wang, R.-Q. Fan, X.-R. Liu, Y.-L. Yang and G.-P. Zhou, J. Inorg. Organomet. Polym., 2012, 22, 744 CrossRef CAS.
- (a) J.-C. G. Bunzli and G. R. Choppin, Lanthanide Probes in Life, Chemical and Earth Sciences: Theory and Practice, Elsevier Scientific Publishers, Amsterdam, 1989 Search PubMed; (b) N. Yuan, T. Sheng, C. Tian, S. Hu, R. Fu, Q. Zhu, C. Tan and X. Wu, CrystEngComm, 2011, 13, 4244 RSC.
- V. Bekiari, G. Pistolis and P. Lianos, Chem. Mater., 1999, 11, 3189 CrossRef CAS.
- S.-R. Zhang, D.-Y. Du, J.-S. Qin, S.-L. Li, W.-W. He, Y.-Q. Lan and Z.-M. Su, Inorg. Chem., 2014, 53, 8105 CrossRef CAS PubMed.
- J. C. Ahern, R. J. Roberts, P. Follansbee, J. McLaughlin, D. B. Leznoff and H. H. Patterson, Inorg. Chem., 2014, 53, 7571 CrossRef CAS PubMed.
- T. J. Sørensen, M. Tropiano, O. A. Blackburn, J. A. Tilney, A. M. Kenwright and S. Faulkner, Chem. Commun., 2013, 49, 783 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: IR spectra, UV-visible spectra, excitation spectra, thermogravimetric analysis (TGA) curves, and solution and solid state NMR spectra. See DOI: 10.1039/c5ra14694a |
| This journal is © The Royal Society of Chemistry 2015 |