
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of porous silicon through interfacial reactions and measurement of its electrochemical response using cyclic voltammetry†
Mingrui
Zhao
a,
Rajesh
Balachandran
b,
Jennie
Allred
b and
Manish
Keswani
*b
aChemical and Environmental Engineering, University of Arizona, Tucson, AZ, USA
bMaterials Science and Engineering, University of Arizona, 1235 E. James E. Rogers Way, Tucson, AZ 85721, USA. E-mail: manishk@email.arizona.edu; Fax: +1-520-621-8059
First published on 8th September 2015
Abstract
Porous silicon, an excellent material with fascinating physical and chemical properties, is usually formed by anodic polarization of single crystalline silicon in HF based solutions. Here, we show fabrication of porous silicon films ∼0.5–250 μm thick consisting of macropores and mesopores using a contactless electrochemical approach, where the silicon substrate is not under any external bias. Pore dimensions and porosity have been characterized by scanning electron microscopy (SEM) while subsequent cyclic voltammetry (CV) investigations delineate the underlying topographical differences between blanket and porous silicon surfaces. Our work not only offers a new scalable means of fabricating porous silicon structures but also questions the reliability of existing theories that depend on localized collection of electronic hole carriers through anodization of silicon for pore formation. We believe our results will open pathways for development of realistic models for porous silicon formation.
Introduction
The formation of porous silicon structures under anodic bias first described by Uhlir and Turner has been employed for many applications in solar cells,1–6 fuel cells,7 biology,8 nanoenergetics,9 microelectromechanical systems (MEMS),10 sensors,11 and photonic crystals.12 It is believed that the electrochemical dissolution of silicon occurs at the pore tips where the electric field is enhanced due to collection of holes in this region while the pore wall remains passivated due to depletion of holes.13–18 The exact mechanism of pore formation is still under debate but it is known that the porous structure is associated with two electrons per dissolved silicon atom and occurs at low values of anodic overpotentials.19 The most commonly used electrolytes for porous silicon formation include aqueous electrolytes (HF–H2O mixtures), organic electrolytes (aqueous HF–organic solvent mixtures),20 oxidizing electrolytes containing an oxidizing agent in DI water but without any fluoride, and mixed electrolytes (e.g. aqueous HF solution with an oxidant).21–23Harraz et al. illustrated the role of nature of organic solvent, HF concentration, and doping level on the morphology and depth of random macropores formed in p-type silicon by its electrochemical anodization in aqueous HF–solvent mixtures.24 Their results revealed that solvents such as dimethyl sulfoxide (DMSO) and dimethyl formamide (DMF) favor macropore formation, acetonitrile (ACN) gives a bilayer of nano/macropores, and aqueous HF or ethanol–HF solutions form nanopores. Lower doping density or higher resistivity increased the pore diameter and decreased the pore spacing and above a critical resistivity (>0.1 Ω cm) only macropores were formed. In another study, the number density of random macropores was found to inversely depend on the resistivity (6–1000 Ω cm) of p-Si (100) samples etched anodically in HF–DMF at a current density of 5.5 mA cm−2.25 The pore diameter and depth increased from 0.9 to 4.5 μm and 17.6 to 53 μm with sample resistivity. Interestingly, the pore diameter was unaffected by the current density while the pore wall thickness decreased with increasing current density. The authors concluded that none of the existing theoretical models in literature satisfactorily described the experimental data of their work. Ponomarev and Levy-Clement proposed that in organic electrolytes, the adsorption of organic molecules on silicon surface can locally affect the silicon dissolution during anodization and affect the morphology of the porous layer.16 Chao et al. used cationic surfactants in aqueous HF–ethanol mixtures to fabricate 400 μm deep pores by anodization of p-type (100) silicon.26 In the presence of surfactants, the pores formed were deeper and more unidirectional. It was hypothesized that the non-polar tail of the amphiphilic surfactant attaches on to the H-terminated hydrophobic silicon and the polar head sticks out in to the solution and affects the local electrostatic properties, electrochemical behavior at the silicon–solution interface and the kinetics of silicon dissolution.
In this work, we are presenting a new contactless electrochemical technique for forming macropores and mesopores in p- or n-type (100) silicon of different resistivities. One side of silicon contacts an aqueous HF–DMSO solution containing Pt mesh electrode while the other side faces an aqueous nickel sulfate/boric acid or potassium ferricyanide solution with Ni or Pt mesh electrode immersed in it, respectively (Fig. 1). The role of boric acid is to act as a buffer and reduce variation in pH of the deposition solution. It also serves to subdue the hydrogen evolution reaction (HER) that may simultaneously occur during nickel deposition. Application of a current density at a suitable value between Pt and Ni (or Pt) electrodes generates porosity in silicon (on HF–DMSO side) with the size of pores determined by the doping density in silicon. Interestingly, the use of aqueous hydrochloric acid as the deposition solution under similar conditions does not yield any pores on silicon. The work provides a novel means of generating porous silicon using a technique where the silicon substrate is not an electrode and does not need to be under an external anodic bias. The results of this study are intriguing as they are contrary to the general understanding of the requirement of electrochemical anodization of the Si (by application of a potential) for pore formation. The main advantage of our technique is that it is scalable since the only contact of the silicon substrate is that with the solutions on the two sides.
 | ||
| Fig. 1 Schematic of the dual compartment PTFE cell used for generating porous silicon. Silicon substrate is free of electrical contacts and reactions are motivated by means of applying a suitable current between the Ni (or Pt) anode and Pt cathode. The anodic and cathodic reactions of silicon dissolution and nickel (or ferricyanide) ion reduction occur continuously and concurrently at the silicon interfaces. | ||
Materials and methods
Formation of porous silicon
The experimental set-up consisted of a two chamber polytetrafluoroethylene cell with a silicon wafer placed in between and sealed using a Kalrez® perfluoroelastomer square-ring. Single side polished p- or n-type (100) silicon wafers with different resistivities were used (8–12 Ω cm and 5 mΩ cm). Prior to each experiment, the silicon surface was pre-cleaned with isopropyl alcohol for 1 min, ammonia–peroxide mixture (1NH4OH (29%)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1H2O2 (30%):5H2O by volume) for 5 min and 0.5% HF solution (by weight) for 1 min with each step followed by deionized (DI) water rinsing. The etchant solution contained 8% HF, 8% water and 84% DMSO (all by weight) or 8% HF and 92% water with or without 0.05 mM 4-(1,1,3,3-tetramethylbutyl)phenyl-polyethylene glycol (Triton® X-100), a non-ionic surfactant, while the deposition solution consisted of nickel sulfate (1 M) and boric acid (0.5 M) or potassium ferricyanide (1 M) or hydrochloric acid (1 or 5 M). Platinum mesh cathode and nickel (or Pt) anode were immersed in the DMSO–HF–water or aqueous HF (with or without Triton® X-100) solution and NiSO4 (or potassium ferricyanide or HCl) solution, respectively. A constant current density of 2–10 mA cm−2 was applied between the anode and the cathode for varying times between 0.5 h and 18 h using a suitable current source (B&K Precision Corporation). High resolution SEM (FEI Inspec-S50) and FESEM (Hitachi S-4800) images of the pores were captured and analyzed using ImageJ® software for characterizing the pore dimensions and surface porosity.
:1H2O2 (30%):5H2O by volume) for 5 min and 0.5% HF solution (by weight) for 1 min with each step followed by deionized (DI) water rinsing. The etchant solution contained 8% HF, 8% water and 84% DMSO (all by weight) or 8% HF and 92% water with or without 0.05 mM 4-(1,1,3,3-tetramethylbutyl)phenyl-polyethylene glycol (Triton® X-100), a non-ionic surfactant, while the deposition solution consisted of nickel sulfate (1 M) and boric acid (0.5 M) or potassium ferricyanide (1 M) or hydrochloric acid (1 or 5 M). Platinum mesh cathode and nickel (or Pt) anode were immersed in the DMSO–HF–water or aqueous HF (with or without Triton® X-100) solution and NiSO4 (or potassium ferricyanide or HCl) solution, respectively. A constant current density of 2–10 mA cm−2 was applied between the anode and the cathode for varying times between 0.5 h and 18 h using a suitable current source (B&K Precision Corporation). High resolution SEM (FEI Inspec-S50) and FESEM (Hitachi S-4800) images of the pores were captured and analyzed using ImageJ® software for characterizing the pore dimensions and surface porosity.
Cyclic voltammetry
The surface of macroporous silicon was coated with nickel using a displacement technique described in ref. 27. Triton® X-100 was used as a wetting agent at 0.05 mM concentration in the solution. The deposition of nickel was carried out using 1 M NiSO4 at a solution pH of 8 and temperature of 60 °C for a period of 60 min and the porous layer was characterized using Energy Dispersive Spectroscopy (EDS) (Thermo Noran System Six EDS) at acceleration voltage of 30 kV. CV was performed using an Interface-1000 potentiostat (Gamry Instruments) on nickel coated blanket and macroporous silicon samples in a solution of 0.5 M potassium carbonate (supporting electrolyte) with or without 2 mM potassium ferricyanide (electroactive species). Potentials were scanned in the range of −0.05 to −0.5 V (versus Hg/Hg2SO4 (sat K2SO4) reference) at a scan rate of 20 mV s−1 (cathodic first). The choice of the supporting electrolyte was based on the stable nickel behavior in K2CO3 solutions compared to the deteriorating performance in commonly used KOH electrolyte.28 Cyclic voltammograms were obtained using two different working electrodes (Pt and Ni) to confirm the positions of the redox peaks (Fig. S1, ESI†).Inductively coupled plasma mass spectrometry (ICP-MS)
DMSO based solutions could not be used for ICP-MS measurements due to compatibility issues between the equipment and the organic solvents. However, since the role of DMSO is mainly to improve silicon wettability, Triton® X-100 surfactant was used in its place in the etchant solution, which allowed measurement of silicon concentration at various times. Aqueous HF (8%) solution w/and w/o the addition of 0.05 mM Triton® X-100 was used as the etchant solution, while the deposition solution still consisted of nickel sulfate (1 M) and boric acid (0.5 M). Pt mesh cathode and Ni plate anode were placed in etchant and deposition solution, respectively. A constant current density of 10 mA cm−2 was applied between the anode and the cathode for 4 h. Samples were drawn from etchant solution every hour and silicon (4+) concentration was analyzed by ICP-MS (Elan DRC-II ICP-MS). SEM images of porous silicon formed in the presence of surfactant in aqueous etchant solution show closer and larger pores and shallower pore depth (Fig. S2(a) and (b), ESI†). In the absence of surfactant, porous silicon formation was not observed (Fig. S2(c) and (d), ESI†).Results & discussion
Fig. 2(a)–(i) show SEM images of macropores formed on p-type silicon of resistivity 8–12 Ω cm (doping ∼ 1.5 × 1015 B atoms cm−3) for reaction times of 0.5, 2, and 18 h at fixed current density of 10 mA cm−2 with deposition solution containing NiSO4 and H3BO3. The images corresponding to these times are denoted as (a)–(c), (d)–(f), and (g)–(i), respectively. The first two images in each column are top views of the macropores and the last image is the cross section of the macroporous layer. The samples with 0.5 h of reaction time had the unpolished silicon surface (root mean square (RMS) roughness 200–500 nm determined using atomic force microscopy) in contact with the etchant solution while the samples with 2 and 18 h of reaction times had the polished silicon surface (RMS roughness < 0.5 nm) facing the etchant solution. The macropores on the polished silicon surface appear more homogeneous than those on the unpolished silicon surface indicating the significance of the initial surface morphology in generating uniform porous silicon. Although, some level of surface irregularity is essential to initiate pore formation, any roughness higher than that adversely affects the uniformity of the surface. It may be noted that during pore formation, Ni is electroplated on the backside Si but the film easily peels off when washed with DI water due to its poor adhesion to Si. | ||
| Fig. 2 SEM images of silicon with macropores from different treatment time and conditions. (a)–(c) 0.5 h of reaction time with unpolished silicon surface in contact with HF–H2O–DMSO etchant solution, (d)–(f) 2 h of reaction time with polished silicon surface in contact with the HF–H2O–DMSO etchant solution, (g)–(i) 18 h of reaction time with polished silicon surface in contact with the HF–H2O–DMSO etchant solution, and (j)–(l) 1 h of reaction time with polished silicon surface in contact with the HF–H2O–DMSO etchant solution. Resistivity of p-type (100) silicon = 8–12 Ω cm. Deposition solution: (a)–(i) aqueous solution of 1 M NiSO4 and 0.5 M H3BO3, (j)–(l) aqueous solution of 1.0 M K3Fe(CN)6. Anode: (a)–(i) nickel and (j)–(l) platinum. Cathode: platinum. Pore diameter, wall thickness and porosity were characterized using the top view micrograph while pore depth was determined using the cross-section view. | ||
SEM of the cross section of the macroporous layer shows that the length of the pores increases from ∼15 μm to 80 μm as the silicon reaction time increases from 0.5 to 2 h. The macropores formed on polished silicon (Fig. 2(f)) are relatively straight and perpendicular to the surface with the diameter of the pore almost constant along the entire pore length. On the contrary, the macropores on the unpolished silicon are not as straight and exhibit variation in size along the length. A statistical analysis was performed to calculate the average diameter (davg) of the top of the pores, average wall thickness (tavg), and porosity (p). The values of davg, tavg, and p were computed to be 1.99 ± 0.47 μm, 0.43 ± 0.12 μm, 68% and 2.29 ± 0.37 μm, 0.38 ± 0.13 μm, 63% respectively for samples with reaction times of 0.5 h and 2 h. With further increase of reaction time to 18 h, the depth of the pores increased to about 250 μm. The pore walls were vertical and straight (Fig. 2(i)) except at the top where they have been etched non-uniformly and a web-like or branched structure is formed. The average growth rate of pores decreases from ∼30 to 15 μm h−1 as the pore depth increases from 15 to 250 μm. At higher pore depths, the mass transport of species inside the pores (which occurs by diffusion) becomes important14 and may explain the observed reduction in the growth rate.
When the deposition solution contained K3Fe(CN)6, the maximum current density that could be applied was ∼3 mA cm−2 for the voltage limit of ∼40 V of the power supply. At this current density and reaction time of 1 h, the average size of the pores and the pore growth rate were 1.40 ± 0.16 μm and ∼10 μm h−1, respectively (Fig. 2(j)–(l)). Although, use of ferricyanide lowers the growth rate of pores due to smaller current density, it may still offer benefits compared to nickel sulfate since it does not leave any film on the backside silicon at the end of the process. The use of hydrochloric acid solution (1 M and 5 M) as the deposition solution for porous silicon formation was also investigated. The maximum current density that could be applied with HCl solution was 2 mA cm−2 and no porous silicon was formed under this condition.
Similar experiments were conducted using n- or p-type (100) silicon of resistivity 5 mΩ cm (doping ∼1.2 × 1019 As atoms cm−3 and 2.2 × 1019 B atoms cm−3), which is about 1000 times lower than that of silicon used in earlier experiments. The polished silicon side was exposed to the aqueous HF–DMSO solution as pore morphology was previously observed to be more uniform with this configuration. The deposition solution contained NiSO4 and H3BO3. The field emission (FE)-SEM images of the top surface and the cross section for n-type silicon at the end of 2 h of reaction time are shown in Fig. 3(a) and (b), respectively. The pore dimension analysis (top-view) of the surface indicated that diameter of the pores was in the range of 5–20 nm, which puts them into the category of mesopores. The pores were randomly distributed and roughly spaced 10 to 25 nm from each other. The cross-sectional view shows that the depth of the mesopores was about 500 nm. On the other hand, when p-type silicon was used under similar processing conditions, pores develop into squares with pyramidal bottoms as opposed to cylinders (Fig. 3(c) and (d)). Although such pore shapes are commonly observed during anisotropic wet etching of silicon in KOH solution, it is unclear as to why these structures are formed with our process.
 | ||
| Fig. 3 FESEM images for different silicon types with mesopores. Micrographs of n-type (a) and (b) and p-type (c) and (d) silicon; resistivity = 5 mΩ cm. Top view (a) and (c) and cross-section view (b) and (d). Polished silicon surface in contact with HF–H2O–DMSO etchant solution. Deposition solution – aqueous solution of 1 M NiSO4 and 0.5 M H3BO3. Reaction time = 2 h. Type of doping in silicon wafer has a significant effect on topography of porous silicon. | ||
Further characterization of the macropores was conducted using a CV technique to understand the topographical characteristics. In order to ensure good electrical response of the silicon surface, it was coated with nickel using a displacement technique prior to the CV measurements. SEM and EDS analysis of the nickel coated silicon were performed to evaluate the efficiency of the deposition process. Fig. 4(a) shows cross sectional SEM image of 80 μm deep porous silicon layer with deposited nickel (see Fig. S3 in ESI† for SEM image of nickel coated 15 μm deep porous silicon layer). The micrographs reveal a uniform layer of nickel even at the bottom of the pores as indicated by the bright spots (Ni being a heavier element appears brighter than Si) suggesting complete coverage of the porous layer with nickel in 1 h of the deposition time. In order to confirm this, mapping of silicon and nickel distribution was performed using EDS. Fig. 4(b) and (c) show distribution of Ni and Si respectively on porous and non-porous regions of the cross-section of the sample (Fig. S4 in ESI† presents distribution map of Ni, Si, and O for 15 μm deep porous layer). It can be observed that the nickel appears everywhere on the inside and the top of the pores but is absent in the region below the pores. The area below the pores is that of the silicon exposed after dicing of the samples and therefore no nickel is expected in this region. The silicon distribution map shows presence of Si exclusively in the region below the pores and also in the porous region and on the top of the pores but to a much smaller extent than in the non-porous region. This is not surprising as the pore walls do not have any nickel and consist of only silicon, which should appear when the porous layer and top surface of pores are mapped. The oxygen distribution map (Fig. 4(d)) shows that top of the pores are oxidized more than the inside of the pores with non-porous silicon region showing the least amount of oxidation. The oxidation (of Ni or Si) is likely induced by air during the storage of the samples for 1 day prior to imaging. The EDS spectrum (Fig. S5, ESI†), obtained by scanning the entire SEM image region and averaging the signal for each element, shows the presence of nickel, silicon, oxygen, aluminium, and carbon peaks. The peaks (excluding the one for aluminium which arises due to stage being made of this material) were fitted with a Gaussian distribution and atomic percent for each element was calculated by applying suitable algorithms and corrections. The atomic percent values for nickel, silicon, oxygen and carbon were computed to be 69.3, 23.3, 3.9, and 3.6 respectively. Nickel content is observed to be the highest and significant indicating that the surface of pores is likely completely covered by nickel. The oxygen and carbon (due to organic contaminants from atmosphere) contents were considerably lower than Ni and Si.
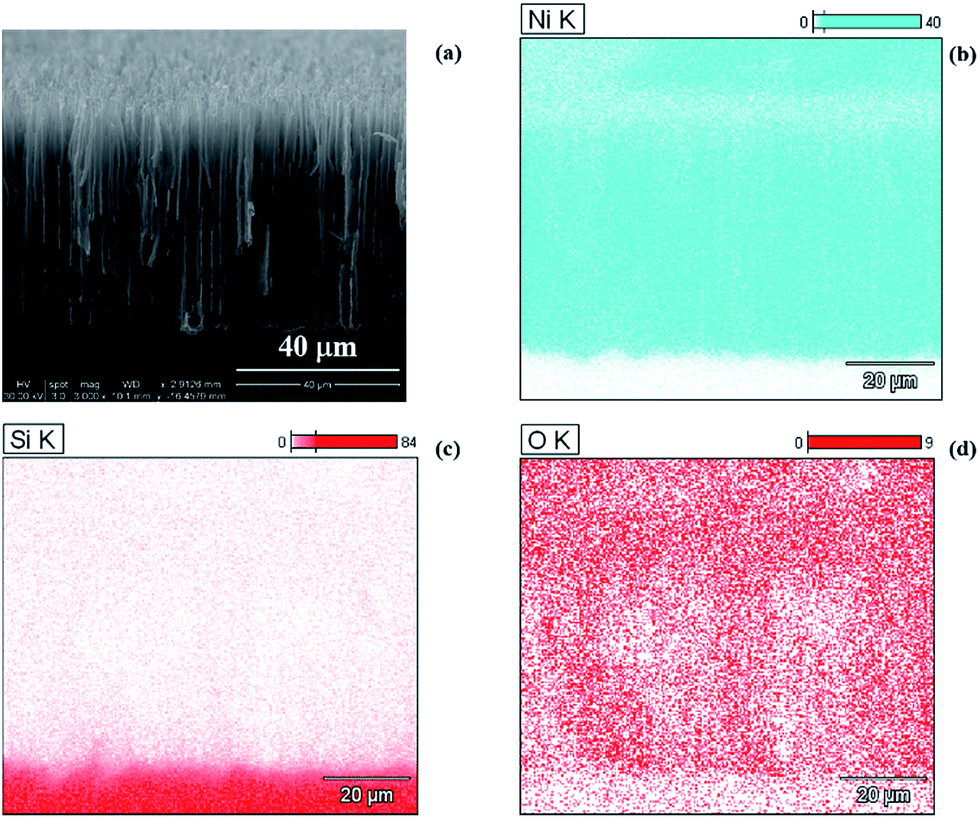 | ||
| Fig. 4 SEM image and EDS distribution mapping for different elements. (a) Cross-section micrograph; (b) nickel, (c) silicon and (d) oxygen distribution maps at cross-section of macroporous Si coated with Ni (80 μm pore depth). Distinguishing feature in the distribution intensity was observed for Ni and Si. Nickel was present only in the porous region while silicon intensity was higher in the non-porous region. | ||
Fig. 5(a) shows the electrochemical response of potassium ferricyanide (with 0.5 M K2CO3 as supporting electrolyte) at nickel coated blanket and porous (80 μm pore depth) silicon samples with projected area of 1 cm2. The current signal for the voltammogram obtained on the blanket electrode is lower than that on the porous silicon electrode. Pair of peaks corresponding to the redox reaction of ferricyanide were observed for the blanket sample with a standard redox potential of −0.17 V (vs. Hg/Hg2SO4) and peak-to-peak potential separation of 84 mV. However, due to large background current in the case of porous silicon (Fig. 5(b)) in comparison to the blanket electrode (up to 15 times higher), the redox peaks were not discernable. Primarily, the significant background current may be attributed to the double layer charging (rearrangement of charged species around the electrode), or redox processes at the large electrode area. Since porous silicon electrode exhibited stable background current, it can be digitally subtracted to reveal changes in current due to ferricyanide redox reaction. The resulting background-subtracted cyclic voltammogram (Fig. 5(c)) indicates that the voltammetric response is improved at the porous electrode as reflected by the enlargement of the oxidation and reduction current peaks. Further, peak-to-peak potential separation is reduced to 52 mV, which may be explained based on the thin layer effects where the solution in the pores becomes depleted of the electroactive species in the time scale of the experiments and results in narrowing of the peak separations.29 The depletion is also reflected by the fact that the increase in current for porous silicon electrode compared to the blanket silicon electrode is not in proportion to the difference in the surface area between the two. In order to ensure that the improvement in the electrochemical response and the decline of the potential difference between the anodic and cathodic peaks was not due to trace level of Triton® X-100 remaining in the pores after nickel coating,30 CV was performed on blanket samples in ferricyanide solutions with and without the surfactant and no measureable difference was observed in the peak positions for the two voltammograms (Fig. S6, ESI†).
 | ||
| Fig. 5 CV on nickel coated blanket and macroporous silicon samples. Cyclic voltammograms obtained using 0.5 M potassium carbonate solutions in the (a) presence and (b) absence of 2 mM potassium ferricyanide for nickel coated blanket and porous (80 μm pore depth) silicon surface, and (c) background-subtracted cyclic voltammograms. Scan rate = 20 mV s−1, projected area: 1 cm2. Better electrochemical response was observed for porous silicon owing to its larger effective surface area. | ||
Silicon in contact with HF–H2O–DMSO solution undergoes a two-step dissolution, the first one being electrochemical oxidation to SiO2 according to reaction (R1) and the second one involves chemical etching of SiO2 as per reaction (R2).31 The role of DMSO is to improve the wettability of silicon surface and prevent any hydrogen bubbles sticking on the surface that are formed in reaction (R1).
| Si + 2H2O = SiO2 + H2 + 2H+ + 2e− | (1) |
| SiO2 + 6HF = 2H+ + SiF62− + 2H2O | (2) |
These reactions indicate that only two of the four silicon electrons participate in the interfacial charge transfer while the other two are accepted by a proton to form hydrogen gas. To maintain charge neutrality in the etchant solution, H+ reduction to hydrogen gas occurs on a Pt mesh cathode immersed in this solution. Gas evolution was visually observed on the Pt mesh as well as on the silicon surface exposed to the etchant solution. The electrons at the silicon–etchant solution interface resulting from first reaction diffuse through the bulk of silicon wafer to be consumed at the interface of silicon–deposition solution. At this later interface, nickel or ferricyanide ions are reduced to zero-valent nickel or ferrocyanide ions (Ni2+ + 2e− → Ni or 2[Fe(CN)6]3− + 2e− → 2[Fe(CN)6]4−), respectively and the deposition solution is depleted of positive charges. The cations are replenished through nickel dissolution or water/ferrocyanide oxidation from/at the anode. Both hydrogen ion reduction and nickel or water/ferrocyanide oxidation are enabled by the applied current between the two electrodes. Using aqueous HCl solution as the deposition solution did not yield any pores on silicon. The possible reason for this could be the poor kinetics of hydrogen evolution reaction on silicon surface.32
Further, ICP-MS measurements were conducted to determine the silicon concentration in the etchant solution as a function of time and verify the proposed mechanism. Table 1 shows the comparison between actual and theoretical concentration of silicon and the faradaic efficiency at different times. The theoretical concentration of silicon was calculated using Faraday's law assuming two out of four electrons from a silicon atom contribute to current (per reaction (R1)) while the remaining two participate in hydrogen gas evolution. A high faradaic efficiency (in the range of 86% to 104%) for different times validates our proposed reaction mechanism.
| Etching time (hour) | Theoretical concentration of silicon (ppm) | Actual concentration of silicon (ppm) | Faradaic efficiency (%) |
|---|---|---|---|
| 0 | 0 | 10 | N/A |
| 1 | 683 | 658 | 96 |
| 2 | 1389 | 1450 | 104 |
| 3 | 2116 | 1950 | 92 |
| 4 | 2868 | 2470 | 86 |
The selective dissolution at the pore tip (initially the tip of a surface irregularity) occurs since the field strength and the current flow are higher at the curved surfaces than those at the flat surfaces.33 Additionally, pore tips are associated with surface defects and weakened bonds, which can increase the silicon dissolution rate due to the sensitivity of electrochemical reactions to the surface curvature. The side walls of the pores remain passivated with hydrogen and are inert to attack by fluoride ions in the absence of any available electronic holes due to similar electronegativity of H and Si and low induced polarization.3
Conclusions
In conclusion, we show evidence of macro- and mesoporous silicon formation using a contactless and scalable approach, which provides a new concept for generating porosity without the need for anodizing silicon. We believe that our work will spur more research to further our understanding of the mechanism of forming porous silicon and enable development of realistic models that will allow accurate prediction of the morphological characteristics of the porous layer based on the processing parameters.Acknowledgements
The authors would like to thank Applied Materials, Inc. for partial support of this work.Notes and references
- F. Dai, J. Zai, R. Yi, M. L. Gordin, H. Sohn, S. Chen and D. Wang, Nat. Commun., 2014, 5, 1–11 Search PubMed.
- P. Granitzer and K. Rumpf, Materials, 2010, 3, 943–998 CrossRef CAS.
- V. Lehmann and U. Gosele, Appl. Phys. Lett., 1991, 58, 856–858 CrossRef CAS.
- A. Uhlir, Bell Syst. Tech. J., 1956, 35, 333–347 CrossRef CAS.
- D. Turner, J. Electrochem. Soc., 1958, 105, 402–408 CrossRef CAS.
- P. Singh, S. Sharma and N. Ravindra, JOM, 2010, 62, 15–24 CrossRef CAS.
- S. Moghaddam, E. Pengwang, Y.-B. Jiang, A. R. Garcia, D. J. Burnett, C. J. Brinker, R. I. Masel and M. A. Shannon, Nat. Nanotechnol., 2010, 5, 230–236 CrossRef CAS PubMed.
- M. M. Orosco, C. Pacholski and M. J. Sailor, Nat. Nanotechnol., 2009, 4, 255–258 CrossRef CAS PubMed.
- C. R. Becker, S. Apperson, C. J. Morris, S. Gangopadhyay, L. J. Currano, W. A. Churaman and C. R. Stoldt, Nano Lett., 2011, 11, 803–807 CrossRef CAS PubMed.
- V. Lehmann, W. Honlein, H. Reisinger, A. Spitzer, H. Wendt and J. Willer, Thin Solid Films, 1996, 276, 138–142 CrossRef CAS.
- R. Angelucci, A. Poggi, L. Dori, G. C. Cardinali, A. Parisini, A. Tagliani, M. Mariasaldi and F. Cavani, Sens. Actuators, A, 1999, 74, 95–99 CrossRef CAS.
- S. John, Phys. Rev. Lett., 1987, 58, 2486–2489 CrossRef CAS PubMed.
- M. J. J. Theunissen, J. Electrochem. Soc., 1972, 119, 351–360 CrossRef CAS.
- V. Lehmann, J. Electrochem. Soc., 1993, 140, 2836–2843 CrossRef CAS.
- V. Lehmann and U. Gruning, Thin Solid Films, 1997, 297, 13–17 CrossRef CAS.
- E. A. Ponomarev and C. Lévy-Clément, Electrochem. Solid-State Lett., 1998, 1, 42–45 CrossRef CAS.
- E. A. Ponomarev and C. Lévy-Clément, J. Porous Mater., 2000, 7, 51–56 CrossRef CAS.
- R. B. Wehrspohn, F. Ozanam and J.-N. Chazalviel, J. Electrochem. Soc., 1999, 146, 3309–3314 CrossRef CAS.
- R. Smith and S. Collins, J. Appl. Phys., 1992, 71, R1–R22 CrossRef CAS.
- S. N. Sharma, R. K. Sharma, G. Bhagavannarayana, S. B. Samanta, K. N. Sood and S. T. Lakshmikumar, Mater. Lett., 2006, 60, 1166–1169 CrossRef CAS.
- V. P. Parkhutik and E. Matveeva, Electrochem. Solid-State Lett., 1999, 2, 371–374 CrossRef CAS.
- M. Christophersen, J. Carstensen and H. Foll, Phys. Status Solidi A, 2000, 182, 45–50 CrossRef CAS.
- H. Foll, M. Christophersen, J. Carstensen and G. Hasse, Mater. Sci. Eng., R, 2002, 39, 93–141 CrossRef.
- F. Harraz, K. Kamada, K. Kobayashi, T. Sakka and Y. Ogata, J. Electrochem. Soc., 2005, 15, C213–C220 CrossRef.
- A. Vyatkin, V. Starkov, V. Tzeitlin, H. Presting, J. Konle and U. Konigh, J. Electrochem. Soc., 2002, 149, G70–G76 CrossRef CAS.
- K. Chao, S. Kao, C. Yang, M. Hseu and T. Tsai, Electrochem. Solid-State Lett., 2000, 3, 489–492 CrossRef CAS.
- C. Xu, X. Zhang, K. Tu and Y. Xie, J. Electrochem. Soc., 2007, 154, D170–D174 CrossRef CAS.
- W. M. Taama, R. E. Plimley and K. Scott, Electrochim. Acta, 1996, 41, 549–551 CrossRef CAS.
- I. Streeter, G. G. Wildgoose, L. Shao and R. G. Compton, Sens. Actuators, B, 2008, 133, 462–466 CrossRef CAS.
- E. Niranjana, B. E. Kumara Swamy, R. Raghavendra Naik, B. S. Sherigara and H. Jayadevappa, J. Electroanal. Chem., 2009, 631, 1–9 CrossRef CAS.
- Z. Patterson, C. Weber, R. Balachandran, R. Gouk, S. Verhaverbeke and M. Keswani, ECS Electrochem. Lett., 2014, 3, D41–D43 CrossRef CAS.
- R. Dominey, N. Lewis, J. Bruce, D. Bookbinder and M. Wrighton, J. Am. Chem. Soc., 1982, 104, 467–482 CrossRef CAS.
- X. Zhang, J. Electrochem. Soc., 2004, 151, C69–C80 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra14592a |
| This journal is © The Royal Society of Chemistry 2015 |