
Real-space characterization of hydroxyphenyl porphyrin derivatives designed for single-molecule devices
Akitoshi Shiotari†
a,
Yusuke Ozakia,
Shoichi Narusea,
Hiroshi Okuyama*a,
Shinichiro Hattaa,
Tetsuya Arugaa,
Takashi Tamakib and
Takuji Ogawab
aDepartment of Chemistry, Graduate School of Science, Kyoto University, Kyoto 606-8502, Japan. E-mail: hokuyama@kuchem.kyoto-u.ac.jp; Fax: +81 75 753 4000; Tel: +81 75 753 3977
bDepartment of Chemistry, Graduate School of Science, Osaka University, 1-1 Machikaneyama-cho, Toyonaka, Osaka 560-0043, Japan
First published on 3rd September 2015
Abstract
Porphyrin derivatives are potential candidates as constituents of functional molecular devices because their electronic levels can be rationally manipulated by chemical modification. In this work, we deposit a porphyrin molecule with a hydroxyphenyl side group on Au(111), which is designed and synthesized as a basic unit for functional single molecule devices, and observe the bonding structure and electronic states with scanning tunneling microscopy (STM). The molecule changes configuration from a monomer to a cluster to a monolayer as the coverage increases, ruled by the H-bonding interaction through the hydroxyphenyl group and the steric repulsion by the isopentoxy groups. The highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) localized in the porphyrin macrocycle are observed at −1.1 and +1.1 eV, respectively, with respect to the Fermi level. We also deposit a para-phenylene-bridged porphyrin array on the surface using the electrospray method, and observe the local density of states along the array.
I. Introduction
Porphyrins and their related macrocycles have rich optical and redox properties, which can be modulated by appropriate metalation. The flexible tunability of their properties means that they are of potential use in molecular electronics applications.1–5 Recently, Tamaki et al.6 reported a methodology for the synthesis of porphyrin arrays with a programmed sequence. Because the individual porphyrin macrocycle possesses a localized valence state of distinct energy levels, it is possible to engineer the distribution of the frontier orbitals along the array, and thereby to rationally design the transport properties through the molecule. The electronic states of the arrayed molecules were studied using optical and electrochemical methods, which revealed the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) levels of each component. However, these versatile techniques are not able to probe the electronic states in a site-resolved way; it is essential to investigate the spatial distribution of the frontier orbitals along the array for the control of the transport properties through the molecules.Scanning tunneling microscopy (STM) is useful to investigate the electronic states and their spatial distribution in molecules on surfaces. The adsorption and self-assembly of porphyrin derivatives on surfaces have been intensively studied, mainly for the understanding of molecule–electrode interactions, which is essential for their application to molecular electronics.7–13 Not only giant molecules,14–16 but also functional molecules, such as molecular switches17,18 and molecular magnets,19 were imaged and characterized at a single-molecule level. In this work, using STM, we image the hydroxyphenyl porphyrin unit and its array which are synthesized as the basis of a molecular rectifier, and characterize the electronic states associated with the transport properties through the molecule.
II. Experimental
The STM experiments were carried out in an ultrahigh-vacuum chamber (USM-1200, Unisoku). An electrochemically etched tungsten tip was used as an STM probe. The STM images were acquired in the constant current mode at 6 K. Single-crystalline Au(111) was cleaned by repeated cycles of Ar+ sputtering and annealing.10-(4-Hydroxyphenyl)-5,15-bis(3,5-diisopentoxyphenyl) freebase porphyrin molecule (FbP; Fig. 1a) and para-phenylene-bridged porphyrin array of Zn porphyrin [(ZnP)3; Fig. 1b] were synthesized according to previous work.6 The FbP molecules were thermally sublimated from a stainless crucible at ∼540 K and deposited onto the surface kept at ∼90 K. We also used an electrospray ionization method (ADCAP vacuum technology, Japan) to deposit nonvolatile (ZnP)3 onto the surface.20 The molecules were dissolved in a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 methanol/toluene (by volume) mixture to give a concentration of 49 μM. The solution in the atmosphere was introduced into a differential pumping chamber at a typical flow rate of 2 μL min−1 through a needle tube with an applied voltage of 1.4 kV. The Au(111) surface was exposed to the ion flux (typical ion current of 10–30 pA) for 5 min at room temperature.
:1 methanol/toluene (by volume) mixture to give a concentration of 49 μM. The solution in the atmosphere was introduced into a differential pumping chamber at a typical flow rate of 2 μL min−1 through a needle tube with an applied voltage of 1.4 kV. The Au(111) surface was exposed to the ion flux (typical ion current of 10–30 pA) for 5 min at room temperature.
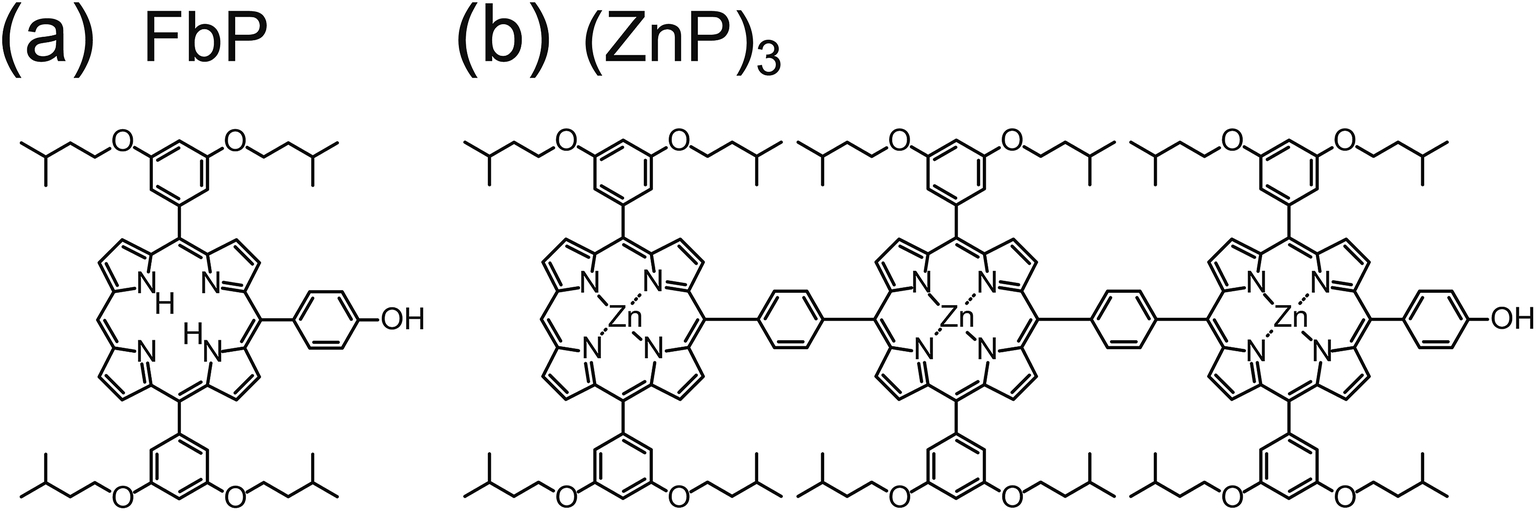 | ||
| Fig. 1 Molecular structures of (a) a freebase porphyrin monomer (FbP) and (b) a phenylene-linked Zn porphyrin array [(ZnP)3]. | ||
The dI/dV curves were obtained using a lock-in amplifier with a modulation voltage of 8 mVrms at 590 Hz. Each spectrum is displayed after subtraction of dI/dV measured over the clean surface at a constant tip height.
III. Results and discussion
A. Porphyrin monomer
Fig. 2 shows STM images of FbP molecules on Au(111) as a function of coverage. The molecules were adsorbed on the surface at ∼90 K. At low coverage (Fig. 2a), the molecules are bonded to the elbow sites of the herringbone structure,21 in a similar way to other porphyrin derivatives on the surface.22,23 The molecule surrounded by the dotted ellipse is an isolated molecule (described below), and clusters are also observed at the elbow sites. In case the surface was exposed to the molecules at room temperature, they were trapped at the step edges on the surface (not shown). The molecules diffused across the surface and migrated to more favorable step sites at room temperature. Therefore it is essential to maintain the surface at ∼90 K to isolate the molecules on the terrace of the surface. As the coverage increases, the molecules form islands in the face-centered-cubic region of the herringbone structure (Fig. 2b), and eventually form a monolayer over the surface (Fig. 2c). | ||
| Fig. 2 (a)–(c) Typical STM images of freebase porphyrin monomers (FbP) on Au(111) as a function of coverage (V = +0.4 V and I = 50 pA). The dotted ellipse in (a) indicates an isolated molecule. The inset in (a) represents the calculated structure of a free FbP molecule (B3LYP/6-31G). The black, blue, red, and white spheres represent C, N, O, H atoms, respectively. | ||
Fig. 3a shows a close inspection into an isolated molecule. The image consists of a ring surrounded by four spots (green dots) and one spot indicated by an arrow. By comparison with the molecular structure (Fig. 3b), we assign the center ring to a porphyrin macrocycle, and the four spots to the isopentoxy groups (green ellipses in Fig. 3b). The arrow indicates the hydroxyphenyl group (yellow ellipse in Fig. 3b). During successive scans, the appearance and position of the four isopentoxy moieties (green dots) change independently, as shown in Fig. 3a, c, and d. The fluctuation is ascribed to the rotation of the isopentoxy moieties around the sp3 C–C bonds, which is induced by the interaction with the STM tip; it depends on the tunnel conditions. We could not suppress the motion under the conditions employed (V = 0.03–2 V and I = 10–500 pA).
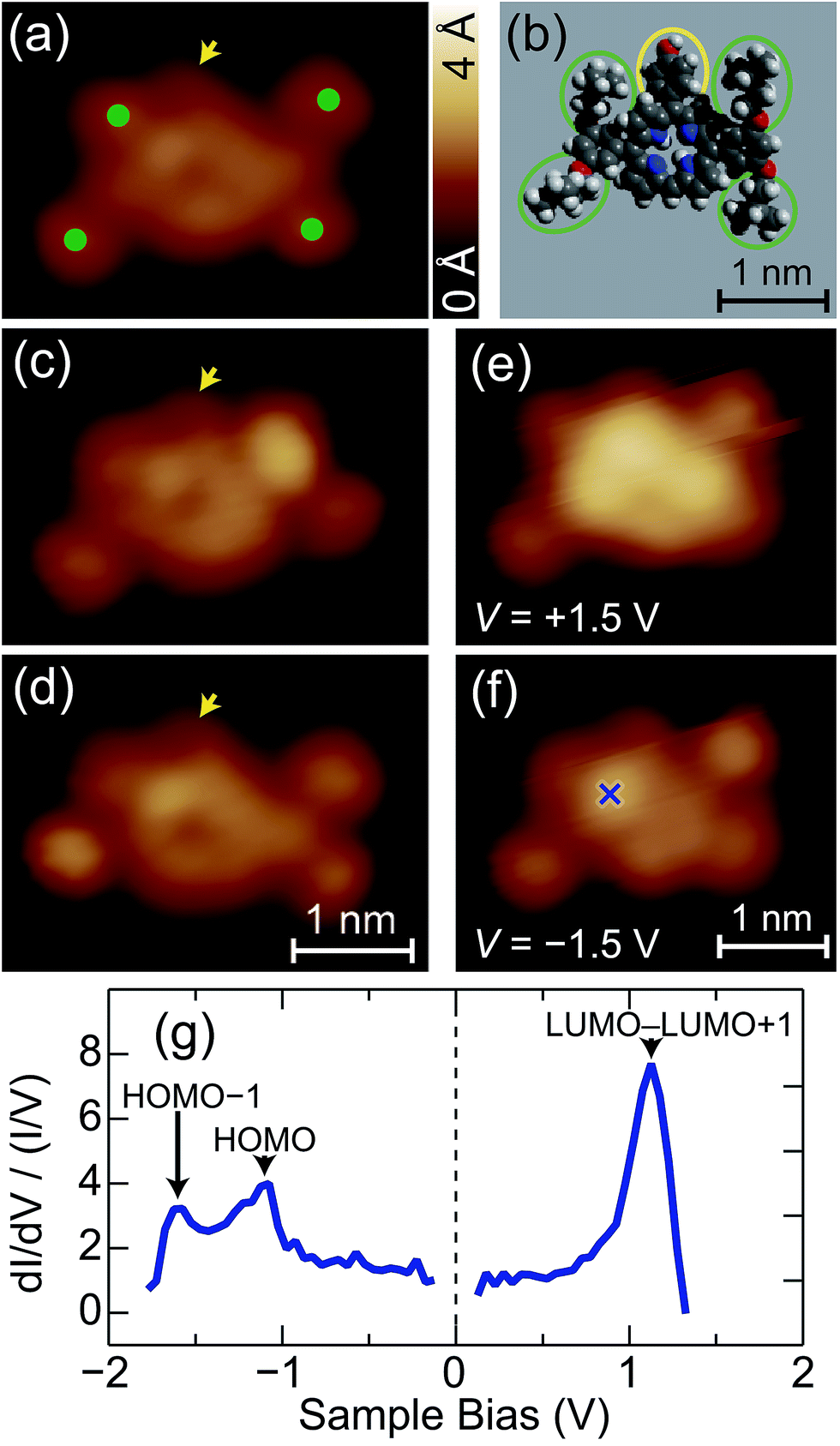 | ||
| Fig. 3 (a) STM images of an isolated FbP molecule (V = +0.4 V and I = 50 pA). The green dots indicate the isopentoxy groups. (b) Schematic illustration of the molecular structure for a FbP molecule. The green and yellow ellipses indicate the isopentoxy and hydroxyphenyl groups, respectively. (c) and (d) STM images of the same molecule as (a), sequentially recorded under the same condition. The arrows in (a), (c), and (d) indicate the hydroxyphenyl group. (e) and (f) STM images of the same molecule as (a), acquired with V = (d) +1.5 and (e) −1.5 V at I = 50 pA. (g) A normalized dI/dV spectrum of the FbP molecule recorded over the cross in (f). The black arrows show the resonance peaks of the molecular orbitals. | ||
Fig. 3e and f show STM images of the molecule obtained at V = +1.5 and −1.5 V, respectively. Depending on the bias voltage, the appearance of the macrocycle drastically changes. It appears as a rather uniform ring at low bias (Fig. 3a), but the upper part of the ring becomes more protruded at V = +1.5 V (Fig. 3e). Note that the upper part of the image corresponds to the site to which the hydroxyphenyl group is bonded (yellow ellipse in Fig. 3b). When the bias polarity is reversed (V = −1.5 V), the bright feature is localized at the specific position of the ring (the cross in Fig. 3f). The dependence of the image on the bias voltage is ascribed to the energy distribution of the electronic states of the molecule. Fig. 3g shows a normalized dI/dV spectrum 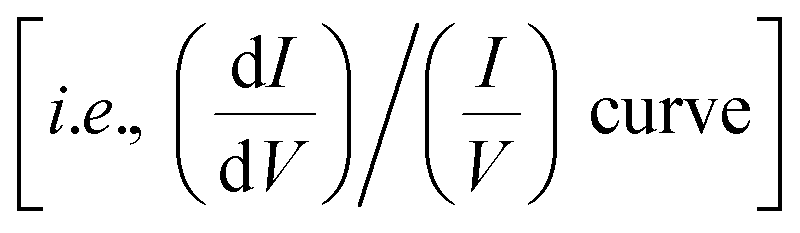 obtained over the cross in Fig. 3f. The density-functional theory (DFT) calculation for the free FbP molecule showed that the LUMO and the second lowest unoccupied molecular orbital (LUMO+1) are distributed within 0.04 eV (ref. 6) and thus may contribute to a peak at V = +1.1 V. The HOMO and the second highest occupied molecular orbital (HOMO−1) are separated from each other by 0.4 eV for the free molecule6 and thus contribute to peaks at V = −1.1 and −1.6 V, respectively. Therefore, the STM images obtained at V = +1.5 V (Fig. 3e) and V = −1.5 V (Fig. 3f) reflect the shape of the LUMO–LUMO+1 and HOMO–HOMO−1 of the molecule, respectively. The HOMO–LUMO gap of ∼2.2 eV is narrower than the calculated value for the free molecule (2.7 eV).6 The reduction of the gap is possibly due to surface polarization effects.24–26
obtained over the cross in Fig. 3f. The density-functional theory (DFT) calculation for the free FbP molecule showed that the LUMO and the second lowest unoccupied molecular orbital (LUMO+1) are distributed within 0.04 eV (ref. 6) and thus may contribute to a peak at V = +1.1 V. The HOMO and the second highest occupied molecular orbital (HOMO−1) are separated from each other by 0.4 eV for the free molecule6 and thus contribute to peaks at V = −1.1 and −1.6 V, respectively. Therefore, the STM images obtained at V = +1.5 V (Fig. 3e) and V = −1.5 V (Fig. 3f) reflect the shape of the LUMO–LUMO+1 and HOMO–HOMO−1 of the molecule, respectively. The HOMO–LUMO gap of ∼2.2 eV is narrower than the calculated value for the free molecule (2.7 eV).6 The reduction of the gap is possibly due to surface polarization effects.24–26
Fig. 4a shows the STM image of dimerized FbP molecules. The molecules preferentially form clusters and islands, as shown in Fig. 2b, suggesting the presence of attractive interactions between the molecules. In Fig. 4a, the molecules in the dimer interact with each other via hydroxyphenyl groups (head-to-head configuration), as illustrated in Fig. 4b. In analogy to similar alkoxyphenyl-substituted cyanophenyl porphyrins,22,27 we attribute the interaction to the H-bonding between the hydroxyl group and the β-hydrogen of the adjacent porphyrin ring. Note that STS performed over the dimerized FbP (not shown) gives the same structure as that of an isolated FbP molecule (Fig. 3g), suggesting that the frontier orbitals are not affected by the H-bonds. At a higher coverage the molecules form a monolayer, as shown in Fig. 4c. On the terrace of the surface, the molecules are arranged in a quasi-rectangular arrangement with the hydroxyphenyl groups pointing in opposite directions at random (Fig. 4d). This is consistent with the β-H⋯OH interaction; the head-to-tail configuration (one H-bond) is nearly degenerate with simultaneous head-to-head (two H-bonds) and tail-to-tail (no H-bond) configurations. Therefore, we suggest that the structure of the monolayer is mainly determined by the interaction between the side isopentoxy groups; the molecules are densely packed by avoiding the steric repulsion between them.
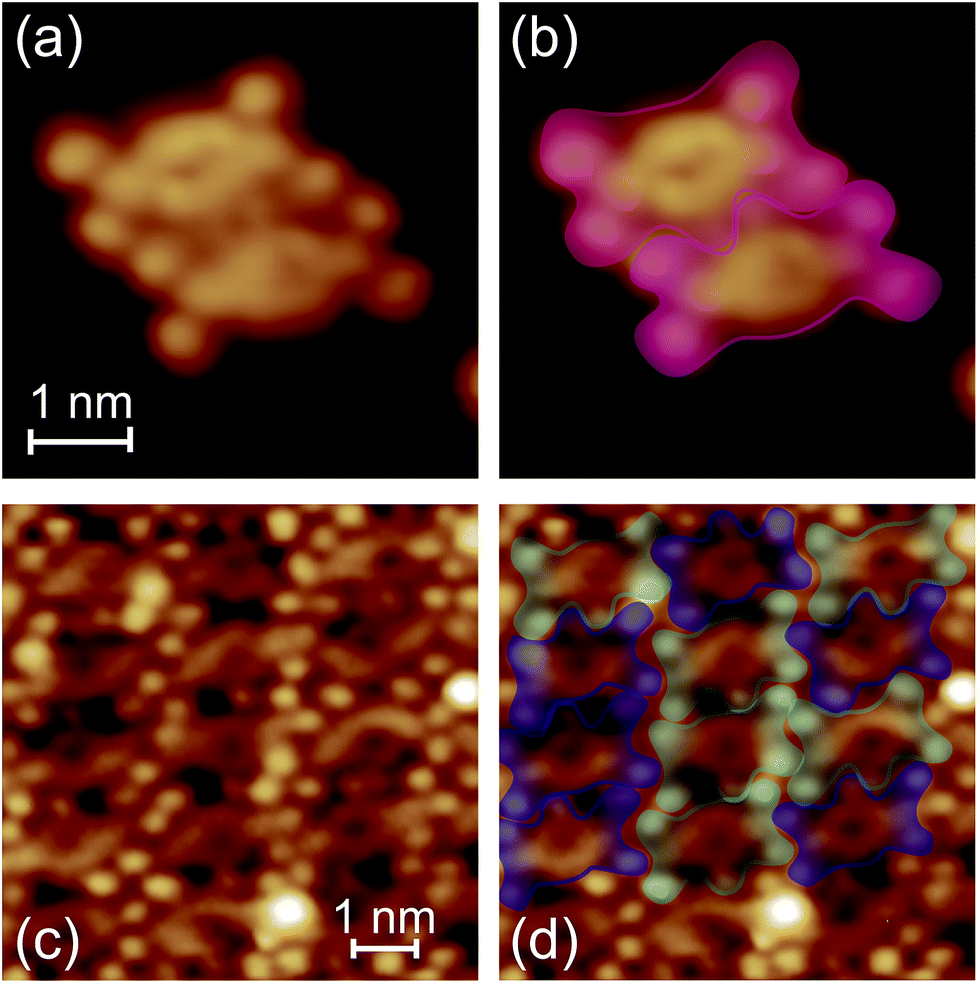 | ||
| Fig. 4 (a) and (c) The STM images of (a) dimerized FbP molecules and (c) monolayer FbP (V = +0.4 V and I = 50 pA). (b) and (d) are the same as (a) and (c), respectively, superimposed by the envelopes representing the contour of the molecules. Blue and cyan envelopes in (d) represent the molecules which are rotated by 180° with respect to each other. | ||
B. Porphyrin array
A variety of metallated porphyrin units can be linked via phenylene in a programmed sequence, enabling one to tailor the transport properties through the molecule.6 Here, as a first step toward the characterization of programmed arrays, we deposit a homologous porphyrin array [(ZnP)3] on the surface and observe the electronic state at the level of individual molecules. Fig. 5a shows typical STM images of (ZnP)3 deposited using the electrospray method on Au(111). Isolated molecules and clusters are observed. Fig. 5b shows a magnified image of an isolated (ZnP)3 molecule. The image is consistent with the molecular structure (Fig. 1b), as illustrated in Fig. 5c. The individual macrocycles with four isopentoxy groups are shown by the red, green, and blue envelopes, corresponding to the head, center, and tail macrocycles, respectively. The residual protrusions (outside of the envelopes) were not reproducible, and thus, are ascribed to impurities attached to the molecule co-adsorbed during the electrospray deposition (possibly solvent molecules).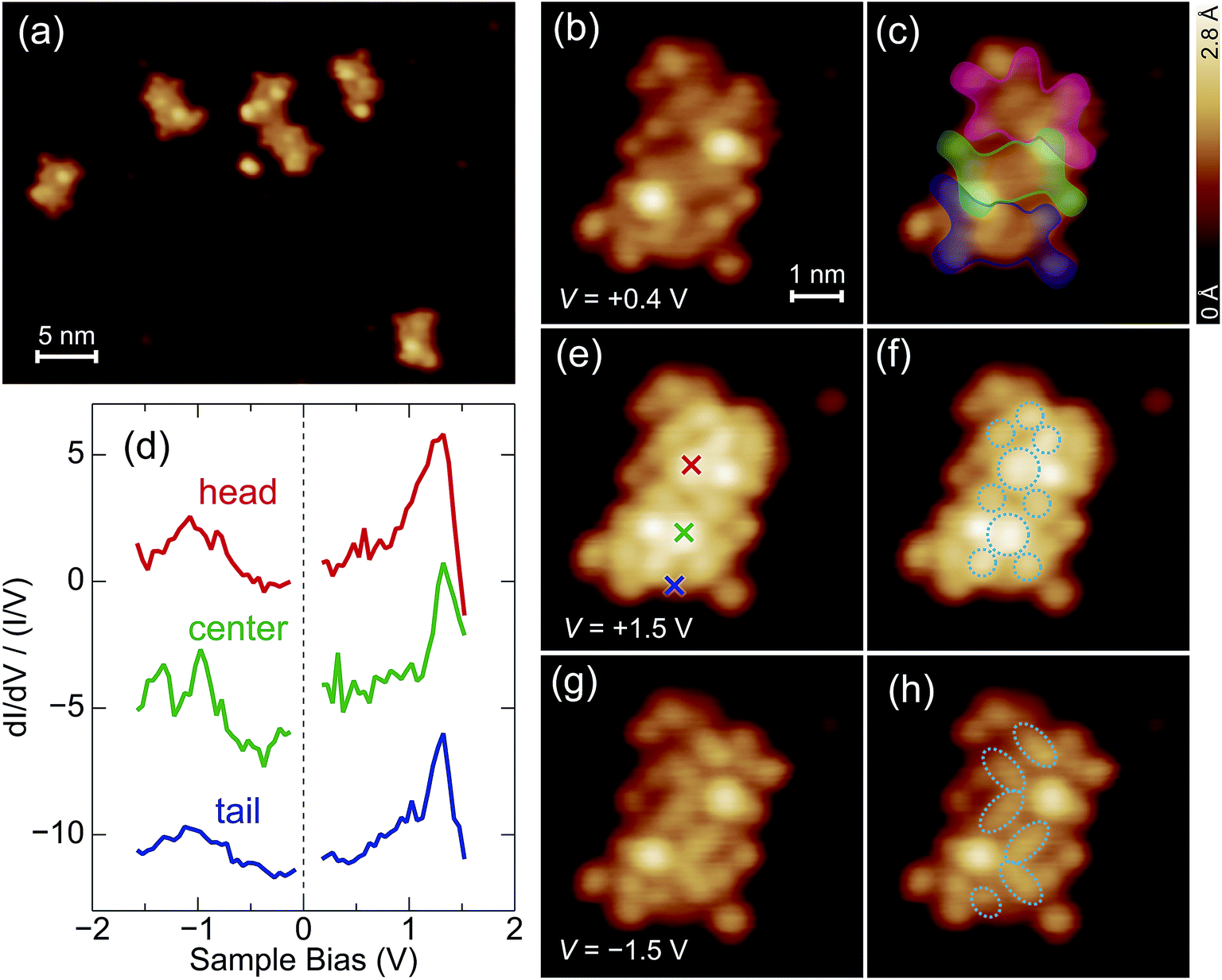 | ||
| Fig. 5 (a) Typical STM images of zinc porphyrin arrays [(ZnP)3] on Au(111) (V = +0.4 V and I = 50 pA). (b), (e) and (g) close-up images of an array acquired with V = (b) +0.4, (e) +1.5, and (g) −1.5 V at I = 50 pA. (c) The same as (b) superimposed by the envelopes representing the contour of each component. (d) Normalized dI/dV curves of the array (offset for clarity). The red, green, and blue curves are recorded over the position marked by the crosses in the corresponding colors in (e). (f) and (h) The same as (e) and (g), respectively, superimposed by the ellipses illustrating the shape of the orbitals distributed in each macrocycle. | ||
The electronic states of (ZnP)3 are investigated with STM as shown in Fig. 5d. The red, green, and blue curves show normalized dI/dV curves obtained over the position marked by the crosses in the corresponding colors in Fig. 5e. The dI/dV curves show the local density of states of the frontier orbitals at each site. As for the unoccupied states, a peak is observed at 1.3 V. The DFT calculation for the free (ZnP)3 molecule showed that five orbitals (LUMO–LUMO+5) are distributed within 0.1 eV (ref. 6) and thus may contribute to the peak. As for the occupied states, the dI/dV curves of the head and tail macrocycles show a broad feature around −1 V, while that of the center macrocycle shows a characteristic split structure. The calculation showed that the HOMO–HOMO−5 orbitals are distributed within 0.3 eV (ref. 6) and thus contribute to these structures. The site-dependence of the dI/dV curves is ascribed to the distribution of so many orbitals along the array. Note that these orbitals may be perturbed by the substrate as well as by the deformation (described later). Therefore DFT calculations for adsorbed (ZnP)3 would help the interpretation of the site-dependent dI/dV curves.
The image obtained at V = +1.5 V (Fig. 5e) has contributions from LUMO–LUMO+5, where the macrocycles show a complex internal structure as depicted in Fig. 5f. The image obtained at V = −1.5 V reflects the shape of the overall HOMO–HOMO−5 (Fig. 5g). The illustration of the occupied-state image (Fig. 5h) shows that each porphyrin macrocycle is no longer symmetric with respect to the molecular axis. The Zn atoms are positioned at the center of the cavities, and thus, we suggest that the asymmetry is due to the deformation of the macrocycles. Indeed, it was suggested that the macrocycle is deformed into a saddle shape with pairs of opposite pyrroles tilted upward or downward to increase the interaction with the surfaces,17,22–24,27–30 which is consistent with the image in Fig. 5h. The two salient protrusions in the molecular image (Fig. 5b) are possibly due to the isopentoxy groups tilting upward according to the deformation of the macrocycles.
IV. Conclusion
We image a freebase porphyrin monomer (FbP) and zinc porphyrin array [(ZnP)3] on Au(111), and characterize their electronic states using STM. The nonvolatile porphyrin array is successfully deposited on the surface using an electrospray deposition. The STM reveals the energy levels and spatial distributions of the frontier orbitals localized on the individual porphyrin macrocycles in the molecules. The real-space imaging is also useful for studying the aggregation on the surface. Combined with a nondestructive deposition method, STM can probe the local electronic states along the programmed porphyrin arrays, which are associated with the transport properties through the molecule.Acknowledgements
This study was supported in part by Grant-in-Aid for Scientific Research on Innovative Areas “Molecular Architectonics: Orchestration of Single Molecules for Novel Functions.” A. S. acknowledges the support of the Japan Society for the Promotion of Science. The authors acknowledge T. Yokoyama for technical support of the electrospray deposition.Reference
- M. Jurow, A. E. Schuckman, J. D. Batteas and C. M. Drain, Coord. Chem. Rev., 2010, 254, 2297–2310 CrossRef CAS PubMed.
- H. Xu, R. Chen, Q. Sun, W. Lai, Q. Su, W. Huang and X. Liu, Chem. Soc. Rev., 2014, 43, 3259–3302 RSC.
- K. Tagami, M. Tsukuda, T. Matsumoto and T. Kawai, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 245324 CrossRef.
- J. A. A. W. Elemans, R. van Hameren, R. J. M. Norte and A. E. Rowan, Adv. Mater., 2006, 18, 1251–1266 CrossRef CAS PubMed.
- G. Sedghi, V. M. García-Suárez, L. J. Esdaile, H. L. Anderson, C. J. Lambert, S. Martin, D. Bethell, S. J. Higgins, M. Elliott, N. Bennett, J. E. Macdonald and R. J. Nichols, Nat. Nanotechnol., 2011, 6, 517–523 CrossRef CAS PubMed.
- T. Tamaki, T. Nosaka and T. Ogawa, J. Org. Chem., 2014, 79, 11029–11038 CrossRef CAS PubMed.
- J. Otuski, Coord. Chem. Rev., 2010, 254, 2311–2341 CrossRef PubMed.
- S. Mohnani and D. Bonifazi, Coord. Chem. Rev., 2010, 254, 2342–2362 CrossRef CAS PubMed.
- S. Yoshimoto and N. Kobayashi, Struct. Bonding, 2010, 135, 137–168 CrossRef CAS.
- J. Xiao, S. Ditze, M. Chen, F. Buchner, M. Stark, M. Drost, H.-P. Steinrück, J. M. Gottfried and H. Marbach, J. Phys. Chem. C, 2012, 116, 12275–12282 CAS.
- T. Niu and A. Li, J. Phys. Chem. Lett., 2013, 4, 4095–4102 CrossRef CAS.
- S. Haq, F. Hanke, J. Sharp, M. Persson, D. B. Amabilino and R. Raval, ACS Nano, 2014, 9, 8856–8870 CrossRef PubMed.
- W. Auwärter, D. Écija, F. Klappenberger and J. V. Barth, Nat. Chem., 2015, 7, 105–120 CrossRef PubMed.
- A. Saywell, J. K. Sprafke, L. J. Esdaile, A. J. Britton, A. Rienzo, H. L. Anderson, J. N. O’Shea and P. H. Beton, Angew. Chem., Int. Ed., 2010, 49, 9136–9139 CrossRef CAS PubMed.
- M. C. O’Sullivan, J. K. Sprafke, D. V. Kondratuk, C. Rinfray, T. D. W. Claridge, A. Saywell, M. O. Blunt, J. N. O’Shea, P. H. Beton, M. Malfois and H. L. Anderson, Nature, 2011, 469, 72–75 CrossRef PubMed.
- D. V. Kondratuk, L. M. A. Perdigao, M. C. O’Sullivan, S. Svatek, G. Smith, J. N. O’Shea, P. H. Beton and H. L. Anderson, Angew. Chem., Int. Ed., 2012, 51, 6696–6699 CrossRef CAS PubMed.
- W. Auwärter, K. Seufert, F. Bischoff, D. Ecija, S. Vijayaraghavan, S. Joshi, F. Klappenberger, N. Samudrala and J. V. Barth, Nat. Nanotechnol., 2012, 7, 41–46 CrossRef PubMed.
- S. Ditze, M. Stark, F. Buchner, A. Aichert, N. Jux, N. Luckas, A. Görling, W. Hieringer, J. Hornegger, H.-P. Steinrück and H. Marbach, J. Am. Chem. Soc., 2014, 136, 1609–1616 CrossRef CAS PubMed.
- W. Wang, R. Pang, G. Kuang, X. Shi, X. Shang, P. N. Liu and N. Lin, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 045440 CrossRef.
- T. Yokoyama, Y. Kogure, M. Kawasaki, S. Tanaka and K. Aoshima, J. Phys. Chem. C, 2013, 117, 18484–18487 CAS.
- J. V. Barth, H. Brune, G. Ertl and R. J. Behm, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 42, 9307–9318 CrossRef CAS.
- T. Yokoyama, S. Yokoyama, T. Kamikado, Y. Okuno and S. Mashiko, Nature, 2001, 413, 619–621 CrossRef CAS PubMed.
- T. Yokoyama, S. Yokoyama, T. Kamikado and S. Mashiko, J. Chem. Phys., 2001, 115, 3814–3818 CrossRef CAS PubMed.
- A. Weber-Bargioni, W. Auwärter, F. Klappenberger, J. Reichert, S. Lefrançois, T. Strunskus, C. Wöll, A. Schiffrin, Y. Pennec and J. V. Barth, ChemPhysChem, 2008, 9, 89–94 CrossRef CAS PubMed.
- J. B. Neaton, M. S. Hybertsen and S. G. Louie, Phys. Rev. Lett., 2006, 97, 216405 CrossRef CAS.
- G. Heimel and J.-L. Bredas, Nat. Nanotechnol., 2013, 8, 230–231 CrossRef CAS PubMed.
- N. Wintjes, J. Hornung, J. Lobo-Checa, T. Voigt, T. Samuely, C. Thilgen, M. Stöhr, F. Diederich and T. A. Jung, Chem.–Eur. J., 2008, 14, 5794–5802 CrossRef CAS PubMed.
- F. Buchner, K. Comanici, N. Jux, H.-P. Steinrück and H. Marbach, J. Phys. Chem. C, 2007, 111, 13531–13538 CAS.
- W. Auwärter, K. Seufert, F. Klappenberger, J. Reichert, A. Weber-Bargioni, A. Verdini, D. Cvetko, M. Dell’Angela, L. Floreano, A. Cossaro, G. Bavdek, A. Morgante, A. P. Seitsonen and J. V. Barth, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 245403 CrossRef.
- T. Yokoyama and F. Nishiyama, J. Phys. Chem. Lett., 2014, 5, 1324–1328 CrossRef CAS PubMed.
Footnote |
| † Present address: Department of Advanced Materials Science, The University of Tokyo, Kashiwa 277-8561, Japan. |
| This journal is © The Royal Society of Chemistry 2015 |