
An immobilized molybdenum acetylacetonate complex on expanded starch for the epoxidation of stillingia oil†
Mei
Hong
,
Meng-Yue
Yao
and
Hui
Pan
*
Jiangsu Key Lab of Biomass-Based Green Fuels and Chemicals, College of Chemical Engineering, Nanjing Forestry University, 159 Longpan Road, 210037, Nanjing, China. E-mail: Hpan@njfu.edu.cn
First published on 12th October 2015
Abstract
An active heterogeneous catalyst, namely a molybdenum acetylacetonate complex immobilized on expanded corn starch (ECS-MoO2(acac)2), was prepared and its catalytic activity for epoxidation of stillingia oil with tert-butyl hydroperoxide (TBHP) was investigated. The heterogeneous catalysts were characterized using inductively coupled plasma optical emission spectrometry, Fourier transform infrared spectroscopy, thermogravimetry and differential thermal analyses, scanning electron microscopy, N2 adsorption–desorption and X-ray photoelectron spectroscopy. By using this catalyst, an environmentally benign process for epoxide production in a heterogeneous manner was developed. The catalyst could be recovered easily and reused without significant degradation in its activity for at least 5 times.
1 Introduction
Stillingia oil is the seed oil of the Chinese tallow tree. It has great potential as an important raw material for the production of oleochemicals because it contains ca. 90% unsaturated compounds and it is a non-edible oil. Use of this oil could lead to a lower consumption of edible oils for the production of chemicals. In addition, tallow trees can grow in most types of soil. The seed output is more than 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 metric tons every year in China.1
000 metric tons every year in China.1
The epoxidation of vegetable oils is commercially important since the epoxides produced from these renewable raw materials present numerous applications, including the fabrication of polyurethane foams (via oxirane ring opening to generate polyols),2 synthetic detergents,3 coatings4 and lubricants.5 The production of biodegradable lubricants from epoxidized vegetable oils is of particular interest considering the undesirable impact on the environment associated with the use of mineral oil-based lubricants.
On an industrial scale, the epoxidation of plant oils is currently carried out with a percarboxylic acid, such as peracetic or performic acid. Soluble mineral acids, commonly sulfuric acid, are used as catalysts for this reaction. Therefore, environmental concerns related to the disposal of the salts formed during the final neutralization of the mineral acid and other technical problems associated with their use, such as corrosion and separation operations, constitute a strong driving force in searching for alternatives to this technology.
The use of transition metal complexes as homogeneous or heterogeneous catalysts could eliminate many of these problems and also provides a reaction that is very selective.6,7 Heterogeneous catalysts have some advantages over homogeneous catalysts, such as the facile separation and recovery of solid catalysts from the reaction mixture for recycling without tedious work.8 Among the transition metal complexes chiefly used as catalysts, molybdenum complexes are currently of considerable interest for promoting oxidation reactions. Several studies in the literature have reported the epoxidation of alkenes using homogeneous6 or heterogeneous Mo(VI) catalysts with tert-butyl hydroperoxide (TBHP) as an oxygen source.9 Recently Farias10 reported the use of molybdenum compounds heterogenized on montmorillonite K-10 for the catalytic epoxidation of soybean and castor oils with TBHP. Epoxidation reactions presenting high yields and selectivity have been observed, as well as no leaching of the metal, with an increase in the catalytic activity for recycling experiments.
Starch is a non-toxic, naturally abundant, biodegradable and highly functional biopolymer which can easily be modified by relatively simple chemical/physical modifications.11–13 However, the application of starch as a material in areas such as composites,14 adsorbents15,16 and catalyst supports17 is restricted by its naturally low surface area (<1 m2 g−1), pore volume (<0.1 cm3 g−1) and low site accessibility. Therefore, expanded starches, which have high surface areas and pore volumes have emerged to be promising materials and their use in catalysis is key for the development of new and improved processes for chemical reactions.18
In the present study, we anchored molybdenum acetylacetonate onto expanded corn starch (ECS) functionalized with 3-aminopropyltriethoxysilane (APTES), through Schiff condensation between the carbonyl groups of the acetylacetonate ligands and the APTES amine. The ability of this heterogeneous catalytic system to catalyze the epoxidation of stillingia oil, using toluene as solvent and TBHP as an oxidizing agent has been investigated (Scheme 1). Reusability is one of the most important properties of a supported heterogeneous catalyst because transition-metal complexes are often expensive and difficult to prepare. Therefore, recycling experiments of the catalyst were also performed in this work.
 | ||
| Scheme 1 The epoxidation of stillingia oil using TBHP as the oxidant. | ||
2 Experimental
2.1 Materials
Degummed stillingia oil was purchased from Dawu county, Hubei province, PR China. The fatty acid composition (based on gas chromatography) is as follows: palmitic acid, 6.1%; stearic acid, 2.2%; oleic acid, 14.9%; linoleic acid, 31.4%; linolenic acid, 44.3% and traces of other fatty acids.Toluene was acquired from Nanjing Chemical Factory (Nanjing, China). Sodium bisulphite and anhydrous sodium sulphate were purchased in analytical grade from Sigma-Aldrich. A solution of anhydrous TBHP in toluene was obtained by careful azeotropic distillation of 70% aqueous solution (Merck) in toluene and the concentration of the resulting solution was determined using 1H NMR spectroscopy. Quantofix® Peroxide 100 test strips (Sigma) were used for the semi-quantitative determination of peroxide. All chemicals were used without further purification. High amylose corn starch and molybdenum acetylacetonate (MoO2(acac)2) were purchased from Merck and used as received. It has been found that higher amylose starches generally give materials with higher surface area after treatment than lower amylose starches and retrograde more rapidly.19
2.2 Catalyst preparation
Expanded corn starch (ECS) was prepared according to a previously reported method.13 In general, high amylose corn starch was initially gelatinized through heating in water at 100 °C for 45 min and the gelatinous mixture was kept at 5 °C for 24 h. Precipitation and washing this gel with ethanol retains a rigid porous gel network with surface areas of >100 m2 g−1.The expanded corn starch is known to possess extensive hydroxyl groups on the framework surface of the porous material. These hydroxyl groups can react with APTES to yield amine functionalized expanded corn starch (ECS-NH2). MoO2(acac)2 was anchored onto the surface of the amine functionalized expanded corn starch (ECS-MoO2(acac)2) through Schiff-base condensation with the free amine groups, covalently attached to the expanded corn starch surface with the carbonyl groups of the acetylacetonate ligand coordinated to molybdenum(VI). Namely, expanded corn starch (4.23 g) was added to a round bottom flask containing dry toluene (35 mL) and stirred under nitrogen. APTES (4.82 g, 22 mmol) was then added and the slurry was refluxed for 24 hours. After this time the vessel was cooled to room temperature and ethanol (135 mL) was added. The resulting ECS-NH2 was recovered by filtration and washed with excess ethanol. The ECS-NH2 (2 g) was then reacted with MoO2(acac)2 (0.30 g, 0.92 mmol) in toluene for 24 h under nitrogen and refluxing conditions. The colored material was then filtered and thoroughly washed with acetone to remove impurities and non-anchored material to yield the final catalyst product ECS-MoO2(acac)2. The preparation steps of the immobilization of MoO2(acac)2 on expanded corn starch are presented in Scheme 2.
 | ||
| Scheme 2 Schematic representation of molybdenum acetylacetonate immobilization onto APTES-functionalized expanded corn starch. | ||
2.3 Catalyst characterization
The molybdenum content of the resulting solid ECS-MoO2(acac)2 was quantitatively determined using inductively coupled plasma optical emission spectrometry (ICP-OES) in a Perkin-Elmer Instruments Optima 2000 DV spectrometer and the loading was found to be 0.37 mmol g−1. The specific surface area (SBET) values of the samples were measured on a Autosorb-iQ instrument through N2 adsorption–desorption isotherms at 77 K using the Brunauer–Emmett–Teller (BET) method. The IR spectra were recorded on a Nicolet 380 FT-IR instrument. A Quanta 200 field emission scanning electron microscope (SEM) was used for the determination of the morphology of the catalyst. Thermogravimetry (TG) and differential thermal analyses (DTA) were carried out with a TGA instruments thermal analyzer TG 209F1 under N2 atmosphere at a heating rate of 10 °C min−1. The X-ray photoelectron spectrometry (XPS) spectra of the catalyst were carried out on a Kratos Axis Ultra DLD spectrometer. 1H NMR spectra were recorded on a Bruker Avance 300 spectrometer.2.4 Epoxidation of stillingia oil
Stillingia oil (1 g; 1.2 mmol; equivalent to 7.2 mmol of double bonds based on 1H NMR) was placed in a 50 mL round bottomed flask connected to a reflux condenser. ECS-MoO2(acac)2 (0.29 g; equivalent to 1.5 mol% of the double bonds present in stillingia oil) and anhydrous TBHP (10.8 mmol; equivalent to 1.5 times of molar number of the double bonds present in stillingia oil) were added to toluene. The mixture was kept under vigorous stir and refluxed for 2 h. At the end of the required reaction time, the catalyst was removed from the reaction mixture by filtration. The filtrate flask was placed into an ice bath, sodium bisulphite solution (15%, w/v) was added slowly and the consumption of peroxide monitored using Quantofix Peroxide 100 test strips. The organic phase was separated, dried over anhydrous sodium sulphate, filtered and the solvent was removed using a rotary evaporator. A small quantity of the sample was analyzed using 1H NMR techniques. 1H NMR spectra of the original and epoxidized stillingia oil are provided in the ESI (S1 and S2).† The conversions of double bonds, epoxide yields and selectivity were calculated based on 1H NMR according to the following equations:6 | (1) |
 | (2) |
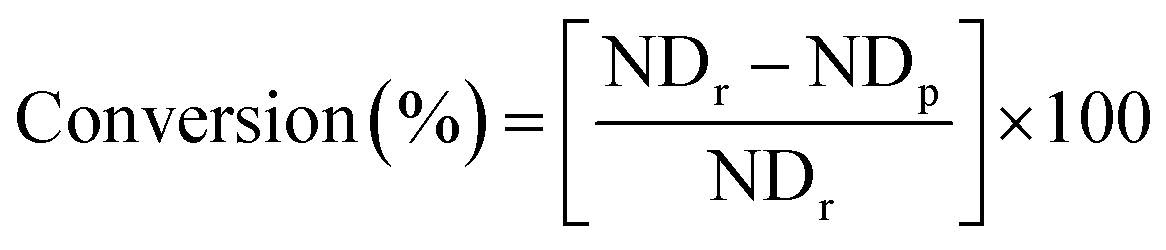 | (3) |
 | (4) |
 | (5) |
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–) and the methine hydrogen of the glycerol moiety, which all appear at the same shift region (5.3–5.4 ppm). NDr is the number of double bonds present in the original stillingia oil, obtained by substitution into eqn (2) of the appropriate peak areas in the spectrum of the original stillingia oil. NDp is the number of double bonds after epoxidation, obtained by substitution into eqn (2) of the appropriate peak areas in the spectrum of the epoxidized oil. I is the peak area associated with the hydrogens of the epoxide groups (–CHOCH–) in the spectrum of the epoxidized oil and occurs at chemical shifts of 2.9–3.1 ppm.20
CH–) and the methine hydrogen of the glycerol moiety, which all appear at the same shift region (5.3–5.4 ppm). NDr is the number of double bonds present in the original stillingia oil, obtained by substitution into eqn (2) of the appropriate peak areas in the spectrum of the original stillingia oil. NDp is the number of double bonds after epoxidation, obtained by substitution into eqn (2) of the appropriate peak areas in the spectrum of the epoxidized oil. I is the peak area associated with the hydrogens of the epoxide groups (–CHOCH–) in the spectrum of the epoxidized oil and occurs at chemical shifts of 2.9–3.1 ppm.20
Recycling tests of the catalyst were carried out as follows: the catalyst was collected from the previous run by filtration and was washed with toluene, dried at 110 °C under reduced pressure for 6 h and subjected to the next catalytic run under the optimized reaction conditions. Reactions using only the ECS as catalyst were also done. All of the above reactions were performed at least three times independently and presented good reproducibility (error < 5%).
3 Results and discussion
3.1 Catalyst characterization
The surface areas of the samples were assessed using N2 adsorption–desorption analysis. The surface area of the corn starch calculated using the BET method was 18 m2 g−1, whereas for ECS it was 135 m2 g−1. The surface area of ECS-NH2 was found to be 52 m2 g−1. In the case of the ECS-MoO2(acac)2 catalyst, the surface area was determined to be 38 m2 g−1. Obviously, the decrease in the surface area of the ECS-MoO2(acac)2 catalyst compared to the ECS material was due to the presence of organic functionalities anchored on the surface of the ECS material.The morphology of the catalyst was studied using SEM images as shown in Fig. 1. The expanded corn starch particles (Fig. 1a) displayed a dispersed and smooth texture. The surface of ECS-MoO2(acac)2 (Fig. 1b) was found to be a fine homogeneous powder with the loaded catalyst clearly visible.
 | ||
| Fig. 1 Scanning electronic microscopy analysis (SEM) of (a) ECS; and (b) ECS-MoO2(acac)2. | ||
The FT-IR spectra of the ECS, ECS-NH2, MoO2(acac)2, and ECS-MoO2(acac)2 samples are presented in Fig. 2. No significant changes were observed in the ECS structure sensitive vibrations after its modification, indicating that the framework of ECS remained unchanged. There was an overall narrowing of the peak width in the 3700–3000 cm−1 range. This could be due to the decrease of the interlayer water content of the ECS, which was a consequence of APTES grafting onto the ECS-OH surface groups. The NH2 stretching band was not observed in Fig. 2b. It could be masked by the broad OH stretching band. A new weak band at 1560 cm−1 for ECS-NH2 was due to the N–H bending vibrations of the primary amines.21 After Schiff condensation, the spectrum of ECS-MoO2(acac)2 showed the appearance of new bands centered at 1605 and 1562 cm−1 which were due to the combinational vibration of the newly formed CN bonds.22 The appearance of the CN bond also confirmed the successful grafting of MoO2(acac)2 onto the APTES functionalized starch through covalent bonds (Scheme 2).
 | ||
| Fig. 2 FTIR spectra for (a) ECS; (b) ECS-NH2; (c) ECS-MoO2(acac)2; and (d) MoO2(acac)2. | ||
The thermal stability of the ECS-MoO2(acac)2 catalyst was determined through thermal analysis. The TGA curves of ECS, ECS-NH2, ECS-MoO2(acac)2 and free MoO2(acac)2 are shown in Fig. 3. The first stage for all of the ECS based materials is a slight mass loss in the TGA curves at temperatures ranging from 50 up to 150 °C which could be ascribed to the removal of physically adsorbed water and solvent trapped in the ECS materials. A further sharp mass loss occurred around 300 °C which was due to the loss of the grafted organic functionality and starch.23 In addition, the immobilized complex ECS-MoO2(acac)2 shows a higher decomposition temperature, similar to the other two ESC based materials rather than that of the free MoO2(acac)2 complex, suggesting that the molybdenum complex was anchored on the ECS support by covalent bonds.
 | ||
| Fig. 3 TGA curves for (a) ECS; (b) ECS-NH2; (c) MoO2(acac)2; and (d) ECS-MoO2(acac)2. | ||
X-ray photoelectron spectroscopy (XPS) spectra of ECS-MoO2(acac)2 demonstrates the presences of molybdenum atoms in the ECS-MoO2(acac)2 materials. The XPS spectrum of ECS-MoO2(acac)2 shows a characteristic Mo 3d5/2 peak with a binding energy (BE) of 232.5 eV in the Mo 3d region (Fig. 4). The Mo 3d3/2 and 3d5/2 peaks are sharp, indicating that Mo existed in the form of Mo6+ on the catalyst surface.24
 | ||
| Fig. 4 High-resolution XPS spectra at the Mo 3d region for (a) ECS-MoO2(acac)2; and (b) ECS-MoO2(acac)2_Used. | ||
3.2 Influence of reaction parameters on the conversion of stillingia oil
“Neat” expanded corn starch was found to be weakly active in the epoxidation of stillingia oil under our reaction conditions. A significant enhancement in the double bonds conversion and epoxide yield of stillingia oil were observed when expanded starch-supported metal acetylacetonate was used as the catalyst (Table 1). Different metal acetylacetonates supported on expanded starch (i.e., ECS-Ni(acac)2, ECS-Cr(acac)3, ECS-VO(acac)2 and ECS-MoO2(acac)2) were also evaluated as catalysts for the epoxidation of stillingia oil. Among all of the catalysts investigated, ECS-MoO2(acac)2 was found to be the most effective catalyst for the epoxidation reaction (Table 1, entry 6). The catalytic activity of ECS-MoO2(acac)2 was compared with MoO2(acac)2 for the epoxidation of stillingia oil under the same reaction conditions (Table 1, entry 2). It showed that the catalytic activities of these two catalysts were very close, suggesting that the grafting of MoO2(acac)2 to the ECS support did not sacrifice its catalytic activity while adding the advantages of being a heterogeneous catalyst. Stillingia oil was also epoxidized with percarboxylic acid generated in situ with hydrogen peroxide as an oxygen donor and glacial acetic acid as an active oxygen carrier in the presence of H2SO4 (Table 1, entry 7). As can be seen from Table 1, the results obtained by using ECS-MoO2(acac)2/TBHP were superior in the epoxide yield and selectivity to those of the H2SO4/H2O2 system. Other reaction parameters for using ECS-MoO2(acac)2 as the catalyst were further investigated.| Entry | Catalyst system | Conversion (%) | Epoxidation (%) | Selectivity (%) | TONb | TOFc (h−1) |
|---|---|---|---|---|---|---|
|
a Reaction conditions: molar ratio of TBHP:oil double bounds:catalyst 150:100:1.5, reactions carried out at 110 °C with toluene, for 2 h.
b TON: total turnover number, moles of epoxide formed per mole of catalyst.
c TOF: turnover frequency which is calculated using the equation [epoxide]/[catalyst] × time (h−1).
d Reaction conditions: molar ratio of acetic acid:H2O2:oil double bounds:catalyst 70:245:70:1, reaction carried out at 65 °C, for 60 min.
|
||||||
| 1 | ECS/TBHP | 8.5 | 5.6 | 65.8 | — | — |
| 2 | MoO2(acac)2/TBHP | 83.6 | 68.3 | 81.7 | 45.5 | 22.8 |
| 3 | ECS-Ni(acac)2/TBHP | 19.3 | 15.7 | 81.3 | 10.5 | 5.2 |
| 4 | ECS-Cr(acac)3/TBHP | 28.4 | 22.4 | 78.7 | 14.9 | 7.5 |
| 5 | ECS-VO(acac)2/TBHP | 32.2 | 25.8 | 80.2 | 17.2 | 8.6 |
| 6 | ECS-MoO2(acac)2/TBHP | 78.5 | 67.1 | 85.5 | 44.7 | 22.4 |
| 7 | H2SO4/H2O2d | 80.9 | 40.7 | 50.3 | 28.5 | 28.5 |
As illustrated in Table 2, the conversion of double bonds increased with increasing the amount of catalyst from 1 mol% to 1.5 mol% (Table 2, entries 1 and 2). A further increase in the amount of catalyst did not improve the double bonds conversion significantly. It is known that the epoxidation reaction is highly influenced by the amount of peroxide used. The conversion could be elevated by introducing a slight excess of TBHP to shift the equilibrium of the reaction to the right-hand side. The conversion to the epoxide product attained a maximum value of 67.1% at the TBHP/double bond molar ratio of 1.5:1. When the molar ratio exceeded 1.5:1, there was a slight decrease in the conversion of double bonds and epoxide yield (Table 2, entry 5).
| Entry | Catalyst loading (mol%) | TBHP/double bonds (molar ratio) | Conversion (%) | Epoxidation (%) | Selectivity (%) | TONb | TOFc (h−1) |
|---|---|---|---|---|---|---|---|
| a Reaction conditions: reactions carried out at 110 °C with toluene, for 2 h. b TON: total turnover number, moles of epoxide formed per mole of catalyst. c TOF: turnover frequency which is calculated using the equation [epoxide]/[catalyst] × time (h−1). | |||||||
| 1 | 1 | 1.5 | 38.6 | 33.0 | 85.4 | 33 | 16.5 |
| 2 | 1.5 | 1.5 | 78.5 | 67.1 | 85.5 | 44.7 | 22.4 |
| 3 | 2 | 1.5 | 78.7 | 67.3 | 85.5 | 33.7 | 16.8 |
| 4 | 1.5 | 1 | 50.4 | 42.4 | 84.1 | 28.3 | 14.1 |
| 5 | 1.5 | 2 | 78.0 | 64.9 | 83.2 | 43.3 | 21.6 |
The effects of reaction time and temperature were also investigated and the results are shown in Table 3. The maximum conversion of double bonds occurred after 2 h of the reaction at the temperature of 110 °C. The double bonds conversion and epoxide yield increased sharply when the reaction temperature increased from 80 °C to 110 °C (Table 3, entries 2 and 3). This result might be due to the loss of water trapped in the expanded corn starch at 110 °C, which could improve the access of the substrates to the active sites.10
| Entry | Temperature (°C) | Reaction time (h) | Conversion (%) | Epoxidation (%) | Selectivity (%) | TONb | TOFc (h−1) |
|---|---|---|---|---|---|---|---|
|
a Reaction conditions: molar ratio TBHP:oil double bounds:catalyst 150:100:1.5, reactions were carried out in toluene.
b TON: total turnover number, moles of epoxide formed per mole of catalyst.
c TOF: turnover frequency which is calculated using the equation [epoxide]/[catalyst] × time (h−1).
|
|||||||
| 1 | 60 | 2 | 48.0 | 33.7 | 70.2 | 22.5 | 11.2 |
| 2 | 80 | 2 | 58.0 | 44.6 | 76.9 | 29.7 | 14.9 |
| 3 | 110 | 2 | 78.5 | 67.1 | 85.5 | 44.7 | 22.4 |
| 4 | 110 | 1.5 | 57.2 | 44.1 | 77.1 | 29.4 | 14.7 |
| 5 | 110 | 2.5 | 83.5 | 67.5 | 80.8 | 45.0 | 22.5 |
3.3 Reusability of the catalyst
Reusability is important for a heterogeneous catalyst. The reusability of the catalyst was examined by carrying out subsequent reaction cycles under the optimized reaction conditions as described previously. The used catalyst was separated from the reaction mixture by filtration, and washed thoroughly with toluene to remove compounds attached to the surface of the catalyst. The catalysts were reused in recycling tests after vacuum-drying overnight at 110 °C. The epoxide yields of the reused catalysts were 67.1%, 66.3%, 65.7%, 64.5% and 62.8%, respectively, when the catalyst was used for 1, 2, 3, 4 and 5 cycles under the optimized reaction conditions. The XPS results showed a similar Mo 3d band profile to that of a freshly prepared catalyst after it was used for five catalytic cycles (denoted as ECS-MoO2(acac)2_Used, Fig. 4b), which confirms the highly stability of the molybdenyl complex during the catalytic process. These results suggested that the catalyst developed in this study can be recycled at least 5 times without significant loss in its catalytic activity. The slight decrease in the conversion may be due to leaching of the active species (less than 0.5%, determined through ICP analysis of the catalyst after the 5th run).4 Conclusion
Molybdenum acetylacetonate covalently anchored onto amine functionalized expanded corn starch was prepared and characterized. The catalytic activity of the catalyst for the epoxidation of stillingia oil was evaluated using TBHP as the oxidant. This heterogeneous catalyst was found to be highly active and selective. Under optimal conditions, the double bonds conversion and epoxide yield of 78.5% and 67.1%, respectively, for stillingia oil were obtained at the refluxing temperature of toluene after 2 h. The heterogeneous catalyst was proven to be easily recoverable and could be recycled at least five consecutive times without a significant loss in catalytic activity. Thus, the properties of being inexpensive, non-toxic, highly active, highly stable, easily recoverable and reusable render this material a potentially valuable catalyst for industrial applications.Acknowledgements
The work was supported by the Natural Science Foundation of Jiangsu Province (China) (No. BK20140969), the open fund of Jiangsu Key Lab of Biomass-based Green Fuels and Chemicals (JSBGFC13006), the Jiangsu Specially-Appointed Professor program of the State Minister of Education of Jiangsu Province and the Specialized Research Fund for the Doctoral Program of Higher Education (G2014006).Notes and references
- L. B. Wen, Y. Wang, D. L. Lu, S. Y. Hu and H. Y. Han, Fuel, 2010, 89, 2267 CrossRef CAS PubMed.
- S. T. Anuar, Y. Y. Zhao, S. M. Mugo and J. M. Curtis, J. Am. Oil Chem. Soc., 2012, 89, 1951 CrossRef CAS.
- Y. Guo, J. H. Hardesty, V. M. Mannari and J. L. Massingill Jr, J. Am. Oil Chem. Soc., 2007, 84, 929 CrossRef CAS.
- M. A. de Luca, M. Martinelli and C. C. T. Barbieri, Prog. Org. Coat., 2009, 65, 375 CrossRef CAS PubMed.
- C. S. Madankar, A. K. Dalai and S. N. Naik, Ind. Crops Prod., 2013, 44, 139 CrossRef CAS PubMed.
- M. Farias, M. Martinelli and D. P. Bottega, Appl. Catal., A, 2010, 384, 213 CrossRef CAS PubMed.
- J. Jiang, Y. Zhang, D. Cao and P. Jiang, Chem. Eng. J., 2013, 215–216, 222 CrossRef CAS PubMed.
- M. D. Serio, R. Turco, P. Pernice, A. Aronne, F. Sannino and E. Santacesaria, Catal. Today, 2012, 192, 112 CrossRef PubMed.
- J. K. Satyarthi and D. Srinivas, Appl. Catal., A, 2011, 401, 189 CrossRef CAS PubMed.
- M. Farias, M. Martinelli and G. K. Rolim, Appl. Catal., A, 2011, 403, 119 CrossRef CAS PubMed.
- A. Shaabani, A. Rahmati and Z. Badri, Catal. Commun., 2008, 9, 13 CrossRef CAS PubMed.
- G. Xie, X. Shang, R. Liu, J. Hu and S. Liao, Carbohydr. Polym., 2011, 84, 430 CrossRef CAS PubMed.
- S. Doi, J. H. Clark, D. J. Macquarrie and K. Milkowski, Chem. Commun., 2002, 2632 RSC.
- R. L. Wu, X. L. Wang, F. Li, H. Z. Li and Y. Z. Wang, Bioresour. Technol., 2009, 100, 2569 CrossRef CAS PubMed.
- L. Huang, C. M. Xiao and B. Chen, J. Hazard. Mater., 2011, 192, 832 CrossRef CAS PubMed.
- Y. Wang, C. Gong, J. S. Sun, H. Gao, S. Zheng and S. Xu, Bioresour. Technol., 2010, 101, 6170 CrossRef CAS PubMed.
- P. Kandylis, A. Goula and A. A. Koutinas, J. Agric. Food Chem., 2008, 56, 12037 CrossRef CAS PubMed.
- M. J. Gronnow, R. Luque, D. J. Macquarrie and J. H. Clark, Green Chem., 2005, 7, 552 RSC.
- J. H. Clark, J. J. E. Hardy, K. Milkowski, F. M. Kerton, A. J. Hunt and F. E. I. Deswarte, World Pat., 2005011836A1, 2005.
- A. O. Bouh and J. H. Espenson, J. Mol. Catal. A: Chem., 2003, 200, 43 CrossRef CAS.
- C. Pereira, S. Patrício, A. R. Silva, A. L. Magalhães, A. P. Carvalho, J. Pires and C. Freire, J. Colloid Interface Sci., 2007, 316, 570 CrossRef CAS PubMed.
- P. Borah, X. Ma, K. T. Nguyen and Y. Zhao, Angew. Chem., Int. Ed., 2012, 51, 7756 CrossRef CAS PubMed.
- S. Kumar, S. L. Jain and B. Sain, Catal. Today, 2012, 198, 204–208 CrossRef CAS PubMed.
- Z. Zhang, B. Liu, K. Lv, J. Sun and K. Deng, Green Chem., 2014, 16, 2762 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra14581c |
| This journal is © The Royal Society of Chemistry 2015 |