
4-Dodecylbenzenesulfonic acid (DBSA) promoted solvent-free diversity-oriented synthesis of primary carbamates, S-thiocarbamates and ureas†
Ali Reza Sardarian * and
Iman Dindarloo Inaloo
* and
Iman Dindarloo Inaloo
Chemistry Department, College of Sciences, Shiraz University, Shiraz 71454, Iran. E-mail: sardarian@shirazu.ac.ir
First published on 3rd September 2015
Abstract
A simple and highly efficient solvent-free method for the conversion of alcohols, phenols, thiols and amines to primary carbamates, S-thiocarbamates and ureas in the presence of 4-dodecylbenzenesulfonic acid (DBSA) as a cheap and green Brønsted acid reagent has been described. All products were obtained in good to excellent yields and characterized using FT-IR, 1H- and 13C-NMR, MS and CHNS techniques.
Introduction
Carbamates, S-thiocarbamates and ureas are an important class of compounds which have been widely used in various areas, including pharmaceuticals, agrochemicals, protection of amino groups in peptide chemistry, and as linkers in combinational chemistry.1 Furthermore, the role of the carbamate linkage has been extensively studied in structurally diverse natural/semi-synthetic molecules against various diseases, such as antibacterial,2a,b anti-diabetic,2c anti-estrogenic,2d anti-cancer,2e,f anti-inflammatory,2g antitumor,2h anti-Alzheimer,2i anti-helminthes,2i anti-osteoporosis,2j anti-progestational,2k anti-malarial,2l antiviral,2m anti-obesity,2n anticonvulsant2o and anti-HIV2p drugs. S-Thiocarbamates and urea derivatives have usually been used as anti-bacterial,3a receptor antagonists,3b anesthetic,3c pesticide,3d antiviral3e and biological receptors inhibitor agents.3f Besides, these compounds are most noted for their use as commercial herbicides.3Although numerous procedures have been developed for the synthesis of N-substituted carbamates, S-thiocarbamates and ureas, they are commercially produced by the phosgenation of primary amines, in which highly toxic phosgene was used.4 More safe substitution reagents have been introduced such as 1,1,1-trichloromethylformate (diphosgene),5 bis-(1,1,1-trichloromethyl) carbonate (triphosgene),6 trichloroacetyl chloride,7 1,1-carbonyldiimidazole (CDI),8 diethyl carbonates (DEC)9 and dimethyl carbonate (DMC).10a
Moreover, various catalytic systems such as SiO2,10b lead(II) nitrate,10c MCM-41-TBD,10d ytterbium triflate (Y(OTf)4),10e acid functionalized ionic liquids,10f supported zirconia catalyst,10g ionic liquids,10h Cs2CO3,10i have been reported for the preparation of carbamates from the reaction of DMC with an amine.
Also several interesting methodologies have been developed for the synthesis of carbamates without the application of phosgene as a starting material such as: (a) the oxidative carbonylation of amines, thiols and alcohols with CO in the presence of transition metal compounds such as Pd, Ru, Zn, Ni and Mn as a catalyst,11 (b) the reductive carbonylation of nitro compounds such as nitrobenzene with alcohols and CO in the presence of Ru, Rh or Pd compounds.12 However, the toxicity of CO and the deactivation of the noble metal catalytic system made this process very uneconomical, (c) conversation of acids to various carbamates in the presence of Me3P and commercially available chloroformates,13 (d) the reaction of CO2, amines and alcohols in the presence of homogeneous metal catalysts (Sn, Ni, Cs, Ir, Ru)14 and heterogeneous catalysts (CeO2, TiO2–Cr2O3/SiO2 and Cu–Fe/ZrO2–SiO2).15
Another strategy for the synthesis of ureas and carbamates is using Hoffmann, Curtius and Lossen rearrangement through an cyanate intermediate and its trapping with alcohols and amines.16–20
Various methods for the synthesis of S-alkyl thiocarbamates have been reported.21 In one of them, condensation of a thiol with a cyanate affords the corresponding S-thiocarbamate. However, this route was only successful if alkoxy and arylsulfonyl cyanates were employed and the application of cyanates, particularly those of low molecular weight, has been avoided due to their high toxicity.7,22 Furthermore, the reaction of primary aliphatic amines with S-alkyl chlorothioformates and then the reaction of thiols with the produced carbamoyl chlorides in the presence of a base afforded the corresponding S-thiocarbamate.7,22a N,N-Disubstituted S-alkyl thiocarbamates have also been prepared from salts of dithiocarbamic acid, which were prepared by the addition of secondary amines to carbon disulfide (CS2).7,22a
Many other reports illustrate the intramolecular rearrangement of various derivatives to afford S-alkyl thiocarbamates. However, these rearrangements are extremely limited in starting substrates.23 Transition metal species containing elements such as palladium, nickel and rhodium have also been employed to promote rearrangement. There are a variety of other methods that are limited to the preparation of complex starting materials.21a,24
Also a two-step synthesis of a variety of S-alkyl thiocarbamate (primary, secondary and tertiary) has been reported via the reaction of thiols with trichloroacetyl chloride followed by the displacement of the trichloroacetyl group upon treatment with an amine.7
There again seems to be only one report in the literature for formation of primary S-thiocarbamates in which the hydration of a variety of organic thiocyanates affords the desired compounds in the presence of hydrogen chloride and concentrated sulfuric acid.7,25
Most of these methods are employed to produce N-substituted carbamates, S-thiocarbamates and ureas and are not able to be used to synthesize N-unsubstituted ones. The use of cyanate salt in the synthesis of primary carbamates through the reaction with alcohols using various kinds of acidic catalysts, such as trichloroacetic acid, silica supported-sulfuric acid, silica supported-perchloric acid and Al(HSO4)3 has been reported.27
4-Dodecylbenzenesulfonic (DBSA), a surfactant that can act as a combined Brønsted acid-surfactant-catalyst (BASC). In the other word, it is able to perform the dual role: (a) as an acid catalyst to activate the substrate molecules and a surfactant to increase the concentration of organic reactants by forming micellar aggregates in water.28 DBSA have been used in chemical synthesis such as synthesis of xanthenes,29,30 bis(indol-3-yl)alkane derivatives,31 carbon–carbon bond forming reactions,32 reaction of homoallyl alcohols and aldehydes in water,33 N-alkylation of aldoximes,34 synthesis of tetrahydrobenzopyrans,35 dehydration reactions in water,36 Thiol-Michael addition reactions,37 1,1-diacetates,38 pyrrolo- and indolo[1,2-a]quinoxalines in ethanol39 and cyclotrimerization of acetophenones.30
Herein, we report a new and highly efficient route for the synthesis of N-unsubstituted carbamates 1, S-thiocarbamates 2 and ureas 3 via the treatment of alcohols, thiols and amines 4 with cyanate salts 5 in the presence of 4-dodecylbenzenesulfonic acid (DBSA) 6 as an effective acid under solvent-free condition (Scheme 1).
 | ||
| Scheme 1 | ||
Results and discussion
For finding a new and suitable preparation method of primary carbamates, the reaction of phenol with potassium cyanate in the presence of DBSA was chosen as the model reaction and optimization of the reaction parameters was performed and the results were summarized in Table 1. According to these data, using molar ratio 1.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1.5 among DBSA, phenol and potassium cyanate respectively under solvent-free and 60 °C gave the highest yield of phenyl carbamate in the shortest time (entry 1, Table 1).
:1:1.5 among DBSA, phenol and potassium cyanate respectively under solvent-free and 60 °C gave the highest yield of phenyl carbamate in the shortest time (entry 1, Table 1).
| Entry | Molar ratio (DBSA:phenol:KNCO) |
Solvent | Temperature (°C) | Time (min) | Yield (%) |
|---|---|---|---|---|---|
| 1 | 1.5:1:1.5 |
— | 60 | 60 | 90 |
| 2 | 1.5:1:1.5 |
— | RT | 180 | 67 |
| 3 | 1:1:1 |
— | 60 | 90 | 78 |
| 4 | 1.2:1:1.2 |
— | 60 | 60 | 84 |
| 5 | 2:1:2 |
— | 60 | 60 | 90 |
| 6 | 1:1:1.5 |
— | 60 | 60 | 75 |
| 7 | 1.5:1:1 |
— | 60 | 60 | 82 |
| 8 | 1.5:1:1.5 |
CH3CN | Reflux | 60 | 63 |
| 9 | 1.5:1:1.5 |
CH3CO2Et | Reflux | 60 | 65 |
| 10 | 1.5:1:1.5 |
CHCl3 | Reflux | 60 | 62 |
| 11 | 1.5:1:1.5 |
CH2Cl2 | Reflux | 60 | 58 |
| 12 | 1.5:1:1.5 |
CCl4 | Reflux | 60 | 50 |
| 13 | 1.5:1:1.5 |
Toluene | Reflux | 60 | 57 |
| 14 | 1.5:1:1.5 |
PEG200 | Reflux | 60 | 48 |
The effect of Brønsted acids such as HCl, H2SO4, CCl3COOH, HClO4, CF3COOH, SiO2–HClO4, SiO2–OSO3H, ClSO3H, p-TSA were explored on the model reaction instead of DBSA. On the basis of results exhibited in Table 2, the reaction was led in the desired product in the presence of all of acids checked but in lower yields after 60 min at 60 °C. Using sodium and ammonium cyanates in the place of potassium cyanate were not conducted to the better yields.
| Entry | Catalyst | Cyanate salt | Yield (%) |
|---|---|---|---|
| a Reaction condition: phenol (1 mmol), MOCN (1.5 mmol), DBSA (1.5 mmol), 60 °C, 60 min. | |||
| 1 | HCl (37%) | KOCN | 25 |
| 2 | H2SO4 | KOCN | 30 |
| 3 | CCl3COOH | KOCN | 70 |
| 4 | HClO4 | KOCN | 62 |
| 5 | CF3COOH | KOCN | 54 |
| 6 | SiO2–HClO4 | KOCN | 60 |
| 7 | SiO2–OSO3H | KOCN | 63 |
| 8 | ClSO3H | KOCN | 56 |
| 9 | p-TSA | KOCN | 55 |
| 10 | DBSA | KOCN | 90 |
| 11 | DBSA | NaOCN | 83 |
| 12 | DBSA | NH4OCN | 80 |
With having the optimized conditions, various alcohols and phenols were treated with potassium cyanate salt in the presence of DBSA at 60 °C to determine efficiency, scope and versatility of the method for one pot and solvent-free synthesis of primary carbamates, which their results were shown in Table 3.
|
||||
|---|---|---|---|---|
| Entry | ROH | Product | Time (min) | Yield (%) |
| a Reaction condition: phenol or alcohol (1 mmol), KOCN (1.5 mmol), DBSA (1.5 mmol), 60 °C. | ||||
| 1 |  |
 |
45 | 83 |
| 2 |  |
 |
45 | 88 |
| 3 |  |
 |
45 | 83 |
| 4 | 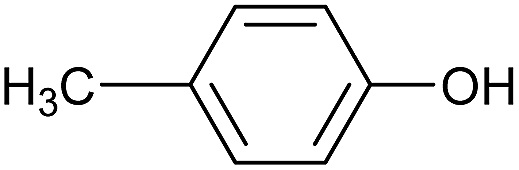 |
 |
45 | 90 |
| 5 |  |
 |
45 | 84 |
| 6 |  |
 |
45 | 90 |
| 7 |  |
 |
60 | 65 |
| 8 |  |
 |
60 | 71 |
| 9 |  |
 |
60 | 70 |
| 10 |  |
 |
60 | 67 |
| 11 |  |
 |
60 | 52 |
| 12 |  |
 |
60 | 65 |
| 13 |  |
 |
60 | 62 |
| 14 |  |
 |
60 | 58 |
| 15 | 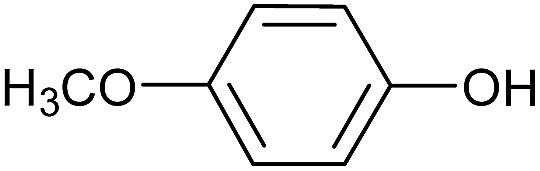 |
 |
30 | 89 |
| 16 |  |
 |
30 | 88 |
| 17 | 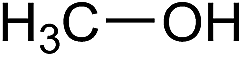 |
 |
30 | 80 |
| 18 |  |
 |
30 | 79 |
| 19 |  |
 |
30 | 85 |
| 20 |  |
 |
30 | 83 |
| 21 |  |
 |
30 | 76 |
| 22 |  |
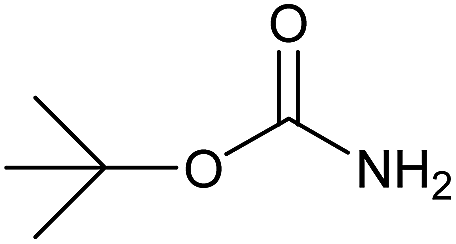 |
30 | 52 |
| 23 |  |
 |
30 | 84 |
| 24 |  |
 |
30 | 87 |
| 25 |  |
 |
30 | 90 |
| 26 |  |
 |
30 | 87 |
| 27 |  |
 |
45 | 82 |
| 28 |  |
 |
45 | 77 |
| 29 |  |
 |
30 | 68 |
| 30 | 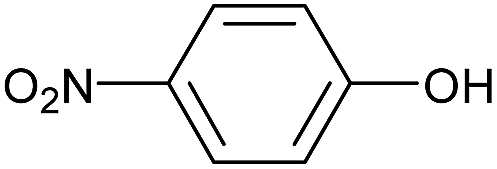 |
 |
60 | NR |
| 31 |  |
 |
60 | NR |
| 32 |  |
 |
60 | NR |
| 33 |  |
 |
60 | NR |

Primary, secondary and tertiary alcohols underwent the carbamate preparation at 60 °C after 30 min in high to moderate yields (Table 3, entries 17–26 and 29) although in the case of tert-butyl alcohol steric hindrance was resulted in lower yield relative to the other class of alcohols (Table 3, entry 22). Phenol and its derivatives with alkyl substitution were also carried out the reaction in high yields after longer reaction time (Table 3, entries 1–7, 27 and 28) except in the case of 2-tert-butyl-4-methylphenol, which suffers from severe steric hindrance of tert-butyl group in the ortho position, which was resulted in lower yield even after longer time (Table 3, entry 7). Phenols with bearing halogen substitutions were also performed the carbamate formation in moderate yields after 60 min (Table 3, entries 8–14) but phenols with strong electron-withdrawing such as NO2, CN, COOR, and CHO (Table 3, entries 30–33) failed to carrying out the reaction under optimized experimental conditions. Most likely these functional groups decrease the nucleophilicity of the phenol oxygen atom for effective attack. On the other hand, phenols with strong electron-donating substitutions, such as methoxy and phenoxy groups were converted to the corresponding carbamates in high yields in shorter reaction times (30 min) and (Table 3, entries 15 and 16). Both isomers of naphthol were also transformed to the desired carbamates after 45 min in 82 and 77% yields (Table 3, entries 27 and 28).
According to the encouraging results obtained from the synthesis of carbamates, it was decided to apply thiols and amines instead of alcohols and phenols for introducing an efficient and facile method for preparation of S-thiocarbamates and ureas.
Therefore, in order to synthesis of primary aliphatic and aromatic S-thiocarbamates, primary aliphatic and aromatic mercaptans were reacted with potassium cyanate in the presence of DBSA and the obtained data are exhibited in Table 4. According to these results, although all of the aliphatic and primary thiols carried out the corresponding S-thiocarbamate in high to excellent yields in relatively short reaction times but aromatic thiols with electron-releasing substitution underwent the reaction in lower yields than aromatic thiols. Among the aliphatic thiols, thiols with steric hindrance produced the desired products in lower yields (Table 4, entries 12 and 13).
|
||||
|---|---|---|---|---|
| Entry | RSH | Product | Time (min) | Yield (%) |
| a Reaction condition: thiols (1 mmol), KOCN (1.5 mmol), DBSA (1.5 mmol), 60 °C. | ||||
| 1 |  |
 |
30 | 93 |
| 2 |  |
 |
30 | 94 |
| 3 |  |
 |
30 | 89 |
| 4 |  |
 |
30 | 85 |
| 5 |  |
 |
30 | 93 |
| 6 | 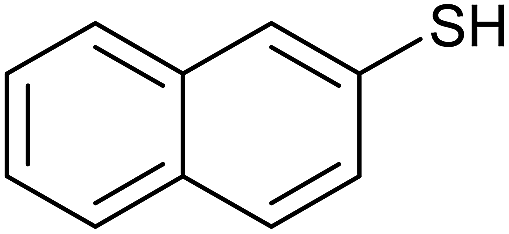 |
 |
30 | 84 |
| 7 |  |
 |
30 | 91 |
| 8 | 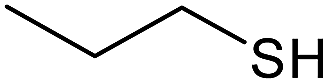 |
 |
30 | 89 |
| 9 |  |
 |
30 | 90 |
| 10 |  |
 |
30 | 92 |
| 11 | 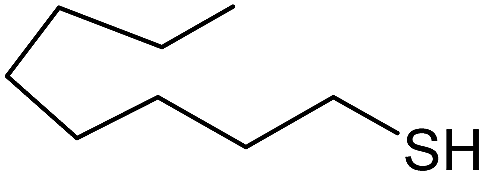 |
 |
30 | 90 |
| 12 |  |
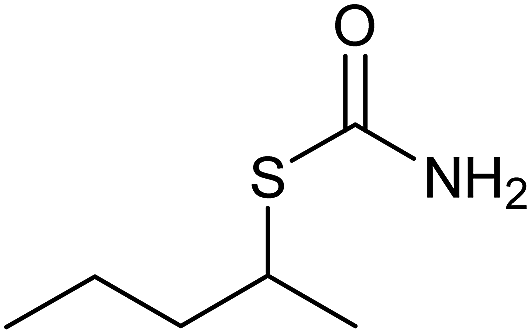 |
30 | 83 |
| 13 |  |
 |
30 | 81 |
| 14 | 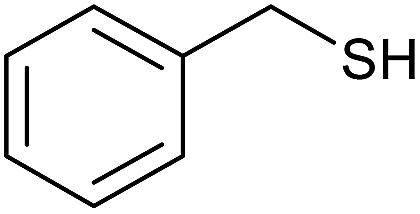 |
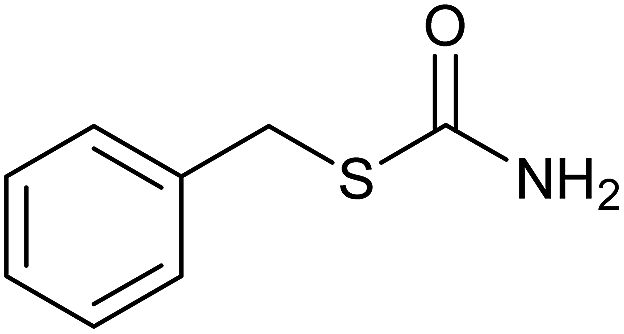 |
30 | 86 |

Preliminary examination on the preparation of ureas from potassium cyanate and primary amines in the presence of DBSA at 60 °C under solvent free conditions, as the same as optimized condition for synthesis of carbamates and S-thiocarbamates, was led to the desired products in lower yields. But raising the reaction temperature up to 80 °C resulted in high to excellent yields. Therefore, variety of primary aromatic amines bearing electron-releasing and electron-withdrawing substituents, primary and secondary aliphatic amines were explored under the new optimized conditions for production of the corresponding ureas, which their information were summarized in Table 5. As the same as for carbamates and S-thiocarbamates, among primary aromatic amine those with electron-donating substitutions underwent the reaction more efficient (Table 5, entries 2 and 4). Except in the case of 4-dimethylaminoaniline, in which dimethylamino group, as an electron-donating substituent, is converted to an electron-attracting one, dimethylammonium salt, in the presence of DBSA (Table 5, entry 5). Aliphatic amines, including primary and secondary, carried out this reaction to provide the title products in high yields (Table 5, entries 7–9).
|
||||
|---|---|---|---|---|
| Entry | RNH2 | Product | Time (min) | Yield (%) |
| a Reaction condition: amines (1 mmol), KOCN (1.5 mmol), DBSA (1.5 mmol), 80 °C. | ||||
| 1 |  |
 |
45 | 90 |
| 2 |  |
 |
45 | 92 |
| 3 |  |
 |
45 | 87 |
| 4 | 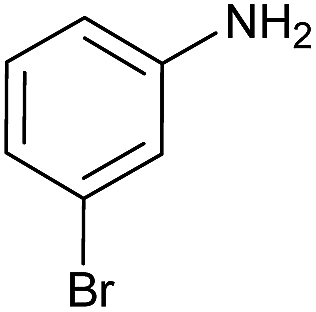 |
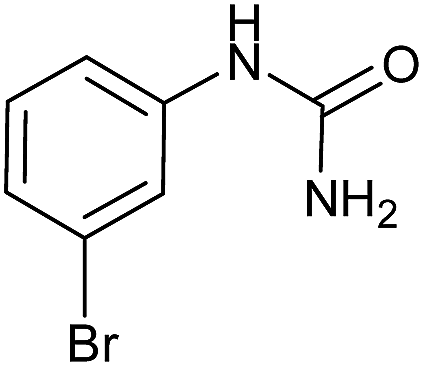 |
45 | 85 |
| 5 |  |
 |
45 | 90 |
| 6 |  |
 |
45 | 81 |
| 7 | 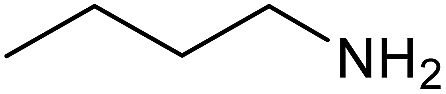 |
 |
45 | 93 |
| 8 |  |
 |
45 | 83 |
| 9 |  |
 |
45 | 86 |

According to the information in the literature26,40 and the results obtained in this study, a plausible mechanism for preparation of the title compounds might be as follows. In the first step, potassium cyanate is protonated by DBSA to produce isocyanic acid that undergoes protonation again and then will be attacked by the existence nucleophile in the media.
According to the plausible mechanism, important features of DBSA, which make it suitable for synthesis of the title compounds, are: (a) its acidic power, which is suitable to produce isocyanic acid and subsequently its protonated form to be attacked by a nucleophile, (b) its viscosity, which prevents escape of isocyanic acid after it is produced therefore rendering a sufficient concentration of isocyanic acid in the reaction media, and (c) its long lipophilic hydrocarbon chain, which provides an organic environment for the reactants.
To checking recognize the applicability of our method at large scales, we examined some reactions in scales of 50 and 200 mmol. The results were summarized in Table 6. As shown in Tables 3–5, the reactions were successfully performed at large scales without significant loss of the yields.
| Product | Phenol, thiophenol or aniline (mmol) | Time (min) | Yield (%) |
|---|---|---|---|
 |
1 | 45 | 83 |
| 50 | 100 | 80 | |
| 200 | 100 | 81 | |
 |
1 | 30 | 93 |
| 50 | 60 | 89 | |
| 200 | 60 | 90 | |
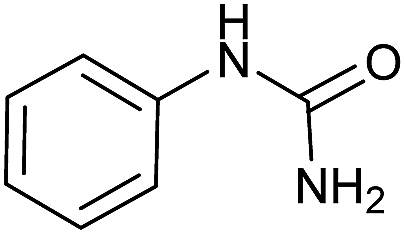 |
1 | 45 | 90 |
| 50 | 100 | 88 | |
| 200 | 100 | 87 |
Experimental
General
All chemicals used in this work were purchased from the Merck, Fluka Chemical Company in high purity. The products were characterized by comparison of their spectral and physical data such as NMR, FT-IR, MS, CHNS and melting point with available literature data. 1H and 13C-NMR spectra were recorded with Bruker Avance DPX 250 MHz instruments with Me4Si or solvent resonance as the internal standard (1H-NMR: Me4Si at 0 ppm, DMSO at δ = 3.35 ppm; 13C-NMR: Me4Si at 0 ppm, DMSO at δ = 39.42 ppm). 1H-NMR spectroscopic data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, quint = quintet, sext = sextet, sept = septet, br. = broad, m = multiplet), coupling constants (Hz), and integration. Fourier transform infrared (FTIR) spectra were obtained using a Shimadzu FT-IR 8300 spectrophotometer. The C, H, N and S elemental analyses were carried out by the using a Thermofinnigan Flash EA-1112 CHNSO rapid elemental analyzer. The mass spectra were recorded on a Shimadzu GC-MS QP 1000 EX instrument. Melting points were recorded by Electrothermal 9100.General procedure for the synthesis of primary carbamates
To a mixture of alcohol or phenol (1.0 mmol) and potassium cyanate (1.5 mmol, 0.121 g) in a mortar, DBSA (1.5 mmol, 0.489 g) was added and the mixture was pulverized until a uniform mixture was formed. Obtained mixture was kept in an oven at a constant temperature (60 °C) for appropriate time (Table 1). During this time, the mixture was grinded for ten seconds every each 5 minutes and the completion of reaction was monitored with TLC using n-hexane:ethyl acetate 9:1 as an eluent. After completion of the reaction, obtained powder was transferred to a beaker, CH2Cl2 (15 mL) and saturated aqueous solution of NaHCO3 (15 mL) were added and organic layer was extracted, washed with water (2 × 15 mL), dried over anhydrous Na2SO4, filtered, and concentrated to afford the crude product. The recrystallization from diethyl ether gave pure product.
General procedure for the synthesis of primary S-thiocarbamates
To a mixture of thiols (1.0 mmol) and potassium cyanate (1.5 mmol, 0.121 g) in a mortar, DBSA (1.5 mmol, 0.489 g) was added and the mixture was pulverized until a uniform mixture was formed. Obtained mixture was kept in an oven at a constant temperature (60 °C) for appropriate time (Table 2). During this time, the mixture was grinded for ten seconds every each 5 minutes and the completion of reaction was monitored with TLC using n-hexane:ethyl acetate 9:1 as an eluent. After completion of the reaction, obtained powder was transferred to a beaker, CH2Cl2 (15 mL) and saturated aqueous solution of NaHCO3 (15 mL) were added and organic layer was extracted, washed with water (2 × 15 mL), dried over anhydrous Na2SO4, filtered, and concentrated to afford the crude product. The recrystallization from diethyl ether gave pure product.
General procedure for synthesis of ureas
To a mixture of amines or anilines (1.0 mmol), and potassium cyanate (1.5 mmol, 0.121 g) in a mortar, DBSA (1.5 mmol, 0.489 g) was added and the mixture was pulverized until a uniform mixture was formed. Obtained mixture was kept in an oven at a constant temperature (80 °C) for appropriate time (Table 3). During this time, the mixture was grinded for ten seconds every each 5 minutes and the completion of reaction was monitored with TLC using n-hexane:ethyl acetate 9:1 as an eluent. After completion of the reaction, obtained powder was transferred to a beaker, CH2Cl2 (15 mL) and saturated aqueous solution of NaHCO3 (15 mL) were added and organic layer was extracted, washed with water (2 × 15 mL), dried over anhydrous Na2SO4, filtered, and concentrated to afford the crude product. The recrystallization from diethyl ether gave pure product.
Conclusion
In conclusion, we have developed a novel and highly efficient method for the synthesis of various primary carbamates, S-thiocarbamates and ureas by the treatment of structurally varied alcohols, phenols, thiols and amines with potassium cyanate in the presence of DBSA, as an efficient, eco-friendly and versatile reagent. The solvent-free conditions, mildness of the conversion, simple experimental procedure, clear reaction profiles, high yields and purity, and short reaction times are the noteworthy advantages of the protocol. Furthermore, this method does not have disadvantages such as involvement of toxic solvents and reagents,4–12,22 expensive starting materials,11–14 formation of any undesirable side products, and epimerization. Further studies are in progress.22,23Acknowledgements
Authors gratefully acknowledge the financial support of this work by the research council of Shiraz University.References
- (a) A. Mateen, S. Chapalamadugu, B. Kashar, A. R. Batthi and G. R. Chaudry, Biol. Degrad. Biorem. Toxic Chem., 1994, 198–233 CAS; (b) D. Chaturvedi, Tetrahedron, 2012, 68, 15–45 CrossRef CAS PubMed; (c) E. T. Harr, R. J. Kowalski, E. Hamel, C. M. Lin, R. E. Longley, S. P. Gunasekera and S. H. Rosenkranz, Biochemistry, 1996, 35, 243–250 CrossRef PubMed; (d) R. J. Kuhr and H. W. Dorough, Carbamate Insecticides: Chemistry, Biochemistry and Toxicology, CRCpress, Cleveland, Ohio, 1976 Search PubMed; (e) Y. Y. Wigfiled, Food Sci. Technol., 1996, 77, 1501–1529 Search PubMed; (f) T. Goto, Y. Ito, S. Yamada, H. Matsumoto, H. Oka and H. Nagase, Anal. Chim. Acta, 2006, 555, 225–232 CrossRef CAS PubMed; (g) D. T. Hung, J. Chen and S. L. Schreibe, Chem. Biol., 1996, 3, 287–293 CrossRef CAS; (h) S. P. Gunasekera, M. Gunasekera and R. E. Longley, J. Org. Chem., 1990, 55, 4912–4915 CrossRef CAS.
- (a) S. J. Shaw, Mini-Rev. Med. Chem., 2008, 8, 276–284 CrossRef CAS; (b) D. K. Hutchinson, Curr. Top. Med. Chem., 2003, 3, 1021–1042 CrossRef CAS; (c) F. Giannessi, P. Pessotto, E. Tassoni, P. Chiodi, R. Conte, F. D. Angeles, N. D. Uomo, R. Catini, R. Deias, M. O. Tinti, P. Carminate and A. Ardnini, J. Med. Chem., 2003, 46, 303–309 CrossRef CAS PubMed; (d) R. Ouellet, J. Rousseau, N. Brasseur, J. E. Lier, M. Doksic and G. Westera, J. Med. Chem., 1984, 27, 509–513 CrossRef CAS; (e) Q. Y. Li, Y. G. Zu, R. Z. Shi and L. P. Yao, Curr. Med. Chem., 2006, 13, 2021–2039 CrossRef CAS; (f) X. Wu and I. Ojima, Curr. Med. Chem., 2004, 11, 429–438 CrossRef CAS; (g) Y. J. Wu and W. J. Su, Curr. Med. Chem., 2001, 8, 1727–1758 CrossRef CAS; (h) Y. Shao, H. Ding, W. Tang, L. Lou and L. Hu, Bioorg. Med. Chem., 2007, 15, 5061–5075 CrossRef CAS PubMed; (i) Y. Ishihara, G. Goto and M. Miyamoto, Curr. Med. Chem., 2000, 7, 341–354 CrossRef CAS; (j) N. David and F. X. Tavares, Curr. Top. Med. Chem., 2005, 5, 1639–1675 CrossRef; (k) L. Zhi, J. D. Ringgenberg, J. P. Edwards, C. M. Tegley, S. J. West, B. Pio, M. Motamedi, T. K. Jones, K. B. Marschke, D. E. Mais and W. T. Schrader, Bioorg. Med. Chem. Lett., 2003, 13, 2075–2078 CrossRef CAS; (l) G. H. Posner, C. H. Oh, L. Gerena and W. K. Milhous, J. Med. Chem., 1992, 35, 2459–2467 CrossRef CAS; (m) L. Garuti, M. Roberti and G. Gentilomi, Farmaco, 2000, 55, 35–39 CrossRef CAS; (n) H.-K. Lee, E. B. Choi and C. Pak, Curr. Top. Med. Chem., 2009, 9, 482–503 CrossRef CAS; (o) B. J. Ludwig and E. C. Piech, J. Am. Chem. Soc., 1951, 73, 5779–5781 CrossRef CAS; (p) P. E. Finke, B. Oates, S. G. Mills, M. MacCoss, L. Malkowitz, M. S. Springer, S. L. Gould, J. A. DeMartino, A. Carella, G. Carver, K. Holmes, R. Danzeisen, D. Hazuda, J. Kessler, J. Lineberger, M. Miller, W. A. Schleif and E. A. Emini, Bioorg. Med. Chem. Lett., 2001, 11, 2475–2479 CrossRef CAS.
- (a) E. L. Ellsworth, T. P. Tran, H. D. H. Showalter, J. P. Sanchez, B. M. Watson, M. A. Stier, J. M. Domagala, S. J. Gracheck, E. T. Joannides, M. A. Shapiro, S. A. Dunham, D. L. Hanna, M. D. Huband, J. W. Gage, J. C. Bronstein, J. Y. Liu, D. Q. Nguyen and R. Singh, J. Med. Chem., 2006, 49, 6435–6438 CrossRef CAS PubMed; (b) M. A. H. Ismail, S. Barker, D. A. El Ella, K. A. M. Abouzid, R. A. Toubar and M. H. Todd, J. Med. Chem., 2006, 49, 1526–1535 CrossRef CAS PubMed; (c) T. Gungor, Y. Chen, R. Golla, Z. P. Ma, J. R. Corte, J. P. Northrop, B. Bin, J. K. Dickson, T. Stouch, R. Zhou, S. E. Johnson, R. Seethala and J. H. M. Feyen, J. Med. Chem., 2006, 49, 2440–2455 CrossRef PubMed; (d) A. W. Erian and S. M. Sherif, Tetrahedron, 1999, 55, 7957–8024 CrossRef CAS; (e) T. F. Wood and J. H. Gardner, J. Am. Chem. Soc., 1941, 63, 2741–2742 CrossRef CAS; (f) M. Huang, W. G. Rice, E. Appella and J. K. Inman, Bioorg. Med. Chem. Lett., 2002, 12, 767–770 CrossRef.
- (a) W. Chen-Hsien, Synthesis, 1981, 622–623 CrossRef; (b) T. Mizuno, I. Nishiguchi, T. Okushi and T. Hirashima, Tetrahedron Lett., 1991, 32, 6867–6868 CrossRef CAS; (c) Y. S. Chen, I. Schuphan and J. E. Casida, J. Agric. Food Chem., 1979, 27, 709–712 CrossRef CAS.
- (a) K. Kurita, T. Matsumara and Y. Iwakura, J. Org. Chem., 1976, 41, 2070–2071 CrossRef CAS; (b) K. Kurita, T. Matsumara and Y. Iwakura, Org. Synth., 1980, 59, 195–201 CAS; (c) K. Kurita, T. Matsumara and Y. Iwakura, J. Org. Chem., 1985, 50, 715–716 CrossRef.
- L. Cotarka, P. Delgu, A. Mardelli and V. Sunjuc, Synthesis, 1996, 553–576 CrossRef.
- J. H. Wynne, S. D. Jensen and A. W. Snow, J. Org. Chem., 2003, 68, 3733–3735 CrossRef CAS PubMed.
- (a) P. A. Duspara, M. S. Islam, A. J. Lough and R. A. Batey, J. Org. Chem., 2012, 77, 10362–10368 CrossRef CAS PubMed; (b) J. A. Grzyb, M. Shen, C. Yoshina-Ishii, W. Chi, R. S. Brown and R. A. Batey, Tetrahedron, 2005, 61, 7153–7175 CrossRef CAS PubMed; (c) V. A. Vaillard, M. González, J. P. Perotti, R. J. Grau and S. E. Vaillard, RSC Adv., 2014, 4, 13012–13017 RSC.
- P. Tundo, C. R. McElroy and F. Aricò, Synlett, 2010, 1567–1571 CrossRef CAS.
- (a) S. Kumar and S. L. Jain, New J. Chem., 2013, 37, 2935–2938 RSC; (b) S. P. Gupta, A. B. Shivarkar and R. V. Chaudhari, J. Chem. Soc., Chem. Commun., 2001, 24, 2620–2621 RSC; (c) T. Baba, M. Fujiwara, A. Oosaku, A. Kobayashi, R. G. Deleon and Y. Ono, Appl. Catal., A, 2002, 227, 1–6 CrossRef CAS; (d) S. Carloni, D. E. De Vos, P. A. Jacobs, R. Maggi, G. Sartori and R. Sartorio, J. Catal., 2002, 205, 199–204 CrossRef CAS; (e) M. Curini, F. Epifano, F. Maltese and O. Rosati, Tetrahedron Lett., 2002, 43, 4895–4897 CrossRef CAS; (f) H. Zhou, F. Shi, X. Tian, Q. Zhang and Y. Deng, J. Mol. Catal. A: Chem., 2007, 271, 89–92 CrossRef CAS PubMed; (g) F. Li, W. Li, J. Li, W. Xue, Y. Wang and X. Zhao, Appl. Catal., A, 2014, 475, 355–362 CrossRef CAS PubMed; (h) A. Zare, T. Yousofi and A. R. Moosavi-Zare, RSC Adv., 2012, 2, 7988–7991 RSC; (i) R. N. Salvatore, S. Shin, A. S. Nagle and K. W. Jung, J. Org. Chem., 2001, 66, 1035–1037 CrossRef CAS.
- (a) S. Fukuoka, M. Chono and M. Kohno, J. Chem. Soc., Chem. Commun., 1984, 6, 399–400 RSC; (b) S. P. Gupte and R. V. Chaudhari, J. Catal., 1988, 114, 246–258 CrossRef CAS; (c) K. T. Li and Y. J. Peng, J. Catal., 1993, 143, 631–634 CrossRef CAS; (d) B. Wan, S. Liao and D. Yu, Appl. Catal., A, 1999, 183, 81–84 CrossRef CAS; (e) W. D. Jones, K. A. Reynolds, C. K. Sperry, R. J. Lachicotte, S. A. Godleski and R. R. Valente, Organometallics, 2000, 19, 1661–1669 CrossRef CAS; (f) H. Kuniyasu, H. Hiraike, M. Morita, A. Tanaka, K. Sugoh and H. Kurosawa, J. Org. Chem., 1999, 64, 7305–7308 CrossRef CAS; (g) A. Bohme and H.-J. Gais, Tetrahedron: Asymmetry, 1999, 10, 2511–2514 CrossRef; (h) J. Jacob, K. A. Reynolds, W. D. Jones, S. A. Godleski and R. R. Valente, Organometallics, 2001, 20, 1028–1031 CrossRef CAS; (i) Y. Nishiyama, H. Kawamatsu and N. Sonoda, J. Org. Chem., 2005, 70, 2551–2554 CrossRef CAS PubMed.
- (a) A. J. Kunin, M. D. Noirot and W. L. Gladfelter, J. Am. Chem. Soc., 1989, 111, 2739–2741 CrossRef CAS; (b) S. J. Skoog, J. P. Campbell and W. L. Gladfelter, Organometallics, 1994, 13, 4137–4139 CrossRef CAS; (c) S. Cenini, C. Crotti, M. Pizzotti and F. Porta, J. Org. Chem., 1998, 53, 1243–1250 CrossRef; (d) F. Ragaini, S. Cenini and F. Demartin, Organometallics, 1994, 13, 1178–1189 CrossRef CAS; (e) E. Alessio and G. J. Mestroni, Mol. Catal., 1984, 26, 337–340 CrossRef CAS; (f) S. Cenini, F. Ragaini, M. Pizzotti, F. Porta, G. Mestroni and E. Alessio, J. Mol. Catal., 1991, 64, 179–190 CrossRef CAS; (g) P. Wehman, G. C. Dol, E. R. Moorman, P. C. Kamer, P. W. van Leeuwen, J. Fraanje and K. Goubitz, Organometallics, 1994, 13, 4856–4869 CrossRef CAS; (h) F. Paul, J. Fischer, P. Ochsenbein and J. Osborn, Organometallics, 1998, 17, 2199–2206 CrossRef CAS; (i) S. T. Gadge and B. M. Bhanage, RSC Adv., 2014, 4, 10367–10389 RSC.
- X. Ariza, F. Urpí and J. Vilarrasa, Tetrahedron Lett., 1999, 40, 7515–7517 CrossRef CAS.
- (a) M. Abla, J. C. Choi and T. Sakakura, Chem. Commun., 2001, 2238–2239 RSC; (b) M. Abla, J. C. Choi and T. Sakakura, Green Chem., 2004, 6, 524–525 RSC; (c) A. Ion, C. Van Doorslaer, V. Parvulescu, P. Jacobs and D. De Vos, Green Chem., 2008, 10, 111–116 RSC; (d) D. F. Niu, L. Zhang, L. P. Xiao, Y. W. Luo and J. X. Lu, Appl. Organomet. Chem., 2007, 21, 941–944 CrossRef CAS PubMed; (e) M. Zhang, X. Zhao and S. Zheng, Chem. Commun., 2014, 50, 4455–4458 RSC; (f) R. A. Watile and B. M. Bhanage, RSC Adv., 2014, 4, 23022–23026 RSC.
- (a) M. Honda, S. Sonehara, H. Yasuda, Y. Nakagawa and K. Tomishige, Green Chem., 2011, 13, 3406–3413 RSC; (b) A. N. Hualiang, L. Zhang, Y. U. A. N. Baoguo, Z. H. A. O. Xinqiang and W. A. N. G. Yanji, Chin. J. Chem. Eng., 2014, 22, 607–610 CrossRef; (c) P. Wang, Y. Ma, S. Liu, F. Zhou, B. Yang and Y. Deng, Green Chem., 2015, 17, 3964–3971 RSC.
- E. W. Wallis and J. F. Lane, Org. React., 1946, 3, 267 Search PubMed.
- (a) R. M. Moriarty, C. J. Chany, R. K. Vaid, O. Prakash and S. M. Tulandher, J. Org. Chem., 1993, 58, 2478–2482 CrossRef CAS; (b) X. Huang and J. W. Keillor, Tetrahedron Lett., 1997, 38, 313–316 CrossRef CAS; (c) X. Huang, M. Seid and J. W. Keillor, J. Org. Chem., 1997, 62, 7495–7496 CrossRef CAS; (d) J. I. Yamaguchi, K. Hoshi and T. Takeda, Chem. Lett., 1993, 1273–1274 CrossRef CAS; (e) Y. Matsumara, T. Maki and Y. Satoh, Tetrahedron Lett., 1997, 38, 8879–8882 CrossRef; (f) G. A. Hiegel and T. Hogenauer, Synth. Commun., 2005, 35, 2091–2098 CrossRef CAS PubMed; (g) T. Nishikawa, D. Urabe, M. Tomita, T. Tsujimoto, T. Iwabuchi and M. Isobe, Org. Lett., 2006, 8, 3263–3265 CrossRef CAS; (h) P. Gogoi and D. Konwar, Tetrahedron Lett., 2007, 48, 531–533 CrossRef CAS PubMed; (i) A. Palmieri, S. V. Ley, K. Hammond, A. Polyzos and I. R. Baxendale, Tetrahedron Lett., 2009, 50, 3287–3289 CrossRef CAS.
- (a) A. A. S. Smith, Org. React., 1946, 3, 337 Search PubMed; (b) E. F. Scriven and K. Turnbull, Chem. Rev., 1988, 88, 297–368 CrossRef CAS.
- (a) L. S. Richer and S. Anderson, Tetrahedron Lett., 1998, 39, 8747–8750 CrossRef; (b) H. Lebel and O. Leogane, Org. Lett., 2005, 7, 4107–4110 CrossRef CAS; (c) H. Lebel and O. Leogane, Org. Lett., 2006, 8, 5717–5720 CrossRef CAS PubMed; (d) O. Leogane and H. Lebel, Synthesis, 2009, 1935–1940 CAS; (e) P. H. Dussault and C. Xu, Tetrahedron Lett., 2004, 45, 7455–7457 CrossRef CAS PubMed; (f) T. Tada, Y. Ishida and K. Saigo, Synlett, 2007, 235–238 CAS; (g) D. Sawada, S. Sasayama, H. Takahashi and S. Iklegami, Tetrahedron Lett., 2006, 47, 7219–7223 CrossRef CAS PubMed; (h) D. Sawada, S. Sasayama, H. Takahashi and S. Iklegami, Tetrahedron, 2008, 64, 8780–8788 CrossRef CAS PubMed; (i) H. L. Yale, Chem. Rev., 1943, 33, 209–256 CrossRef CAS; (j) L. Bauer and O. Exner, Angew. Chem., Int. Ed. Engl., 1974, 13, 376–384 CrossRef PubMed; (k) F. Hamon, G. Prie, F. Lecornue and S. Papot, Tetrahedron Lett., 2009, 50, 6800–6802 CrossRef CAS PubMed.
- P. Dube, N. F. F. Nathel, M. Vetelino, M. Couturier, C. L. Aboussafy, S. Pichette, M. L. Jorgensen and M. Hardink, Org. Lett., 2009, 11, 5622–5625 CrossRef CAS PubMed.
- (a) W. Walter and K. D. Bode, Angew. Chem., Int. Ed. Engl., 1967, 6, 281–293 CrossRef CAS PubMed; (b) A. Waldner and A. Mesmaeker, Synlett, 1995, 108–110 CrossRef CAS; (c) L. J. Zimmerman, H. L. Valentine and W. M. Valentine, Chem. Res. Toxicol., 2004, 17, 258–267 CrossRef CAS PubMed; (d) A. Klasek, V. Mrkvicka, A. Pevec and J. Kosmrlj, J. Org. Chem., 2004, 69, 5646–5651 CrossRef CAS PubMed; (e) R. Hosseinzadeh, M. Tajbakhsh and N. Aghili, Heteroat. Chem., 2015, 26, 175–182 CrossRef CAS PubMed.
- (a) E. Artuso, G. Carvoli, I. Degani, R. Fochi and C. Magistrisa, Synthesis, 2007, 1096–1102 CAS; (b) M. Bejia, H. Sbihi, A. Baklouti and A. Cambon, J. Fluorine Chem., 1999, 99, 17–24 CrossRef.
- R. P. Mull, J. Am. Chem. Soc., 1955, 77, 581–583 CrossRef CAS.
- (a) J. N. Harvey, J. Jover, C. L. J. Guy, J. D. Moseley, P. Murray and J. S. Renny, Angew. Chem., Int. Ed., 2009, 48, 7612–7615 CrossRef CAS PubMed; (b) M. Burns, G. C. Lloyd-Jones, J. D. Moseley and J. S. Renny, J. Org. Chem., 2010, 75, 6347–6354 CrossRef CAS PubMed; (c) G. C. Lloyd-Jones, J. D. Moseley and J. S. Renny, Synthesis, 2008, 661–689 CAS; (d) C. Zonta, O. De Lucchi, R. Volpicelli and L. Cotarca, Top. Curr. Chem., 2007, 275, 131–161 CrossRef CAS; (e) A. Mondragón, I. Monsalvo, I. Regla and I. Castillo, Tetrahedron Lett., 2010, 51, 767–770 CrossRef PubMed.
- (a) X. Zhang and S. Lu, Synlett, 2005, 1535–1538 CAS; (b) A. Klasek, V. Mrkvicka, A. Pevec and J. Kosmrlj, J. Org. Chem., 2004, 69, 5646–5651 CrossRef CAS PubMed.
- (a) B. Movassagh and M. Soleiman-Beigi, Monatsh. Chem., 2008, 139, 137–140 CrossRef CAS; (b) P. Salehi, M. A. Zolfigol, F. Shirini and M. Baghbanzadeh, Curr. Org. Chem., 2006, 10, 2171–2189 CrossRef CAS.
- (a) A. R. Modarressi-Alam, M. Rostamizahed and P. Najafi, Turk. J. Chem., 2006, 30, 269–276 Search PubMed; (b) A. R. Modarressi-Alam, M. Nasrollahzaden and F. Khamooshi, ARKIVOC, 2007, XVI, 238–245 Search PubMed; (c) A. R. Modarressi-Alam, F. Khamooshi, M. Nasrollahzaden and H. A. Amirazizi, Tetrahedron, 2007, 63, 8723–8726 CrossRef; (d) A. R. Modarressi-Alam, M. Nasrollahzaden and F. Khamooshi, Sci. Iran., 2008, 15, 452–455 Search PubMed; (e) R. C. Weast and D. R. Lide, Handbook of Chemistry and Physics, CRC, Florida, FL, 70th edn, 1989, C-194 Search PubMed.
- (a) J. J. Shrikhande, Res. J. Chem. Environ., 2007, 11, 82–83 CAS; (b) D. Prasad, A. Preetam and M. Nath, RSC Adv., 2012, 2, 3133–3140 RSC; (c) J. Li and X. Liu, CrystEngComm, 2014, 16, 184–190 RSC.
- T. S. Jin, R. Q. Zhao and T. S. Li, ARKIVOC, 2006, 11, 176–182 Search PubMed.
- D. Prasad, A. Preetam and M. Nath, C. R. Chim., 2012, 15, 675–678 CrossRef CAS PubMed.
- Y. Y. Peng, Q. L. Zhang, J. J. Yuan and J. P. Cheng, Chin. J. Chem., 2008, 26, 2228–2232 CrossRef CAS.
- K. Manabe, Y. Mori and S. Kobayashi, Tetrahedron, 2001, 57, 2537–2544 CrossRef CAS.
- T. Hatakeyama, Y. Chisaka and C. Kuroda, Helv. Chim. Acta, 2009, 92, 1333–1340 CrossRef CAS PubMed.
- S. K. Hota, A. Chatterjee, P. K. Bhattacharya and P. Chattopadhyay, Green Chem., 2009, 11, 169–176 RSC.
- T. S. Jin, R. Q. Zhao and T. S. Li, ARKIVOC, 2006, 11, 176–182 Search PubMed.
- K. Manabe, S. Lumura, X. M. Sun and S. Kobayashi, J. Am. Chem. Soc., 2002, 124, 11971–11978 CrossRef CAS.
- Y. Chai, D. Dong, Y. Ouyang, Y. Liang, Y. Wang, M. Li and Q. Liu, Lett. Org. Chem., 2007, 4, 281–284 CrossRef CAS.
- M. Esmaeilpour and A. Z. Sardarian, Iran. J. Sci. Technol., 2013, 37A3, 277–284 Search PubMed.
- A. Preetam and M. Nath, RSC Adv., 2015, 5, 21843–21853 RSC.
- (a) A. R. Modarresi-Alam, I. D. Inaloo and E. Kleinpeter, THEOCHEM, 2012, 1024, 156–162 CAS; (b) R. Riemschneider, F. Wojahn and G. Orlick, J. Am. Chem. Soc., 1951, 73, 5905–5907 CrossRef; (c) R. Riemscuneider and G. Orlick, Monatsh. Chem., 1953, 84, 313–318 CrossRef; (d) R. Riemschneider, J. Am. Chem. Soc., 1956, 78, 844–847 CrossRef CAS.
- T. L. Davis and K. C. Blanchard, J. Am. Chem. Soc., 1929, 51, 1790–1801 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of FT-IR, 1H- and 13C-NMR, MS spectra for all compounds. See DOI: 10.1039/c5ra14528g |
| This journal is © The Royal Society of Chemistry 2015 |