DOI:
10.1039/C5RA14486H
(Paper)
RSC Adv., 2015,
5, 99133-99142
Insights into the Morita–Baylis–Hillman reaction of isomeric dibenzofuran carbaldehydes: a theoretical and mass spectral study†
Received
22nd July 2015
, Accepted 30th October 2015
First published on 30th October 2015
Abstract
We herein report the faster Morita–Baylis–Hillman (MBH) reaction of dibenzofuran-4-carbaldehyde (2) than that of its isomer, dibenzofuran-2-carbaldehyde (1), with different activated olefins in the presence of DABCO as the base catalyst. We observed that there is no significant effect of the solvent (methanol) on the reaction rates. In situ mass spectrometry experiments and computational studies were applied to understand the role of the reaction intermediates and their structure implications. MS data revealed that the zwitterionic intermediate obtained from 2 is more stable than that obtained from 1. Computational studies were performed for the gas as well as solvent phase reactions at the mPW1K/6-31 + G(d,p) level. In accordance with the experimental results, aldehyde 2 is found to be more reactive compared to 1. The results are in accordance with McQuade's proposal of the MBH mechanism, wherein the second equivalent of aldehyde plays a key role in the proton migration step during the course of the reaction in the absence of methanol solvent.
Introduction
The Morita–Baylis–Hillman (MBH) reaction1 is one of the most powerful named reactions in organic synthesis for the construction of C–C bonds.2 It is synthetically equivalent to the addition of a vinyl anion to an electrophile (carbonyl compound) in the presence of a tertiary amine as the base catalyst. Contributions to the development of the MBH reaction can be enhanced several fold by modifying the electrophile, activated olefin or catalyst.3 Despite a plethora of reports on the application of MBH adducts, the general sluggishness of the reaction has been a prime concern. The remaining key drawback of the MBH reaction is its poor reaction rate. The attempted methods using ultrasound,4 microwaves,5 molten salts,6 ionic liquids,7 organocatalysis8 and other advanced techniques did not greatly improve the rate of the reaction. All these observations have prompted researchers worldwide to attempt numerous mechanistic investigations9 to reduce the reaction time.
Although studies involving the correlation of theoretical and experimental investigations provide some insights into the MBH reaction, there are many questions to be answered. Enhancing the rate of this reaction is still a puzzle to be solved. The best known faster MBH reactions are those which involve heterocyclic aldehydes. The reaction between substituted 2-chloronicotinic aldehydes10 and methyl acrylate or acrylonitrile completes within 15 min. A similar reaction time was also observed in the case of 3-aryl-5-isoxazolecarboxaldehyde (Fig. 1).11,12 In view of these significant observations, we examined the positional and steric effects of a formyl group on the MBH reaction with isomeric dibenzofuran aldehydes.
 |
| | Fig. 1 Heterocyclic aldehydes previously used as substrates in the Morita–Baylis–Hillman reaction. | |
We herein report our results on the remarkable difference in the rates of MBH reactions of isomeric dibenzofuran aldehydes, i.e., dibenzofuran-2-carbaldehyde (1) and dibenzofuran-4-carbaldehyde (2) with activated olefins in the presence of a base catalyst. The mechanistic aspects of the rate of MBH reaction with aldehydes 1 and 2 were examined using high resolution electrospray ionization-mass spectrometry (ESI-MS), tandem mass spectrometry (MS/MS) and computational studies.
Results and discussion
To start, the required dibenzofuran-2-carbaldehyde (1) was synthesized from dibenzofuran using the procedure developed in our lab.13 Dibenzofuran-4-carbaldehyde (2) was prepared in 60% yield by reacting dibenzofuran with n-butyllithium/dimethylformamide at −78 °C. Both aldehydes 1 and 2 were characterized using NMR and mass spectral data and were correlated with the literature.13,14
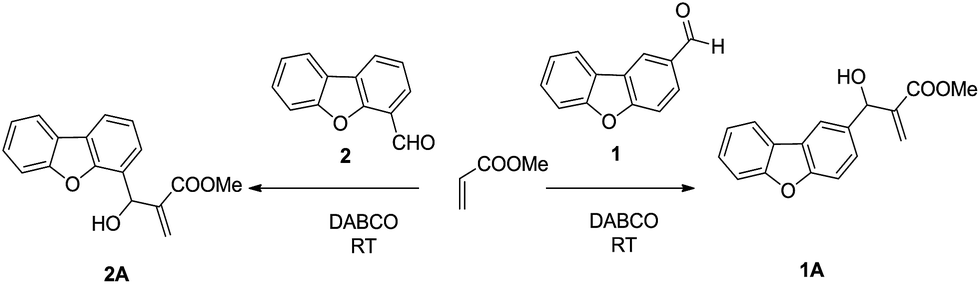
Initiating the study, we performed Morita–Baylis–Hillman (MBH) reactions of aldehydes 1 and 2 with methyl acrylate in the presence of DABCO as a base catalyst (Scheme 1) at room temperature. The reaction with aldehyde 1 took seven days to afford MBH adduct 1A in 75% yield. However, when the same reaction was performed with the isomeric aldehyde 2 under identical reaction conditions, surprisingly, it proceeded to complete conversion within 24 h to give MBH adduct 2A in 99% yield. The products 1A and 2A were fully characterized by 1H NMR, 13C NMR and mass spectral data.
 |
| | Scheme 1 MBH reaction of isomeric aldehydes 1 and 2 with methyl acrylate. | |
Furthermore, we performed a systematic HPLC analysis of the reaction of aldehydes 1 and 2 with methyl acrylate at different reaction time points. Samples of the reaction mixtures drawn at different time intervals (3, 6, 9, 12, 24, 48 and 72 h) were analyzed by HPLC and the percentages of MBH adducts 1A and 2A formed were calculated (Fig. 2). Correlation of the %product yields with reaction time revealed that the MBH reaction between aldehyde 2 and methyl acrylate completed within 24 h to produce adduct 2A, whereas at this reaction time only 13% of adduct 1A was formed with aldehyde 1 (Fig. 2). These results clearly revealed that the MBH reaction of aldehyde 1 was much slower than that of isomeric aldehyde 2.
 |
| | Fig. 2 Percentage of formation of MBH adducts 1A and 2A from the respective reactions of isomeric aldehydes 1 and 2 with methyl acrylate at different time intervals, analyzed by HPLC. | |
Effect of electrophile
In order to obtain further insight, we next examined the MBH reaction with various electrophiles. The isomeric aldehydes 1 and 2 were thus reacted with different electrophiles, such as methyl acrylate, ethyl acrylate, acrylonitrile and 2-cyclohexenone, in the presence of the base, DABCO (Scheme 2) under identical reaction conditions.
 |
| | Scheme 2 MBH reactions of isomeric aldehydes 1 and 2 with various activated olefins A–D. The reaction times and yields are shown in parentheses. | |
All the reactions produced the corresponding MBH adducts 1A–D and 2A–D from the respective isomeric aldehydes 1 and 2; however, their yields varied with the reaction time. The maximum yields of MBH adducts 1A–D and 2A–D with respect to reaction time are presented in Fig. 3. All the MBH adducts 1A–D and 2A–D were fully characterized by 1H NMR, 13C NMR and mass spectral data.
 |
| | Fig. 3 Graphical representation of the maximum yields of MBH adducts 1A–D and 2A–D and their reaction times [aldehyde 1 or 2 (1 mmol), activated olefin (8 mmol) and DABCO (1 mmol) were stirred at room temperature]. | |
The above mentioned experimental results (Fig. 3) revealed that the MBH reactions with aldehyde 2 completed with shorter reaction times than the reactions with the isomeric aldehyde 1. Among the four electrophiles, the reaction with acrylonitrile was faster than the reactions with the other three olefins (Fig. 3). We also observed that all the reactions proceeded with six fold faster rates with aldehyde 2 than with aldehyde 1. This suggests that the differences in the reaction times remained almost the same for both the isomeric aldehydes, irrespective of the electrophile used.
Effect of base and solvent
With a view to verifying the effect of the catalyst (base) on the MBH reaction, we performed the reaction in the presence of different bases. We have chosen the reaction of aldehyde 1 with acrylonitrile for this study to monitor the effect of the base. The MBH reaction (Scheme 3) was performed using different bases such as DBU, quinuclidine, Et3N, imidazole, K2CO3, PPh3 and DMAP. Among all the bases screened, DABCO was found to be the base that gave the best yield (93%) (Table 1). This suggests a key role of DABCO in the differential rates of the MBH reaction of the isomeric aldehydes. Furthermore, the reaction of aldehyde 1 with acrylonitrile in methanol in the presence of DABCO did not show any significant improvement in the rate of BH reaction compared to the blank (without solvent) reaction.
 |
| | Scheme 3 MBH reaction of aldehyde 1 with acrylonitrile and different bases. | |
Table 1 Effect of base on the MBH reaction of aldehyde 1 depicted in Scheme 3
| S. No. |
Base |
Yielda (%) |
| All the reactions were performed at room temperature and yields were reported at 22 h. n.r = no reaction was observed. |
| 1 |
DABCO |
93 |
| 2 |
DBU |
<25 |
| 3 |
Et3N |
n.rb |
| 4 |
K2CO3 |
n.rb |
| 5 |
DMAP |
<20 |
| 6 |
PPh3 |
<10 |
| 7 |
Imidazole |
n.rb |
| 8 |
Quinuclidine |
23 |
Mechanistic studies using in situ ESI-MS analysis
Over the past decade, ESI-MS has become an important tool15 in mechanistic studies because of the soft ionization process that occurs in ESI-MS; moreover, it produces intact molecule ions directly from the solution phase. In the present study, we used ESI-MS to investigate the mechanism of the MBH reaction of isomeric dibenzofuran aldehydes. Tandem mass spectrometry (MS/MS) and high resolution mass spectrometry (HRMS) techniques were used for the characterization of key reaction intermediates. Based on the results discussed in the previous sections, we selected the MBH reaction of the isomeric aldehydes 1 and 2 with methyl acrylate in the presence of DABCO for detailed mass spectral analysis.
According to the widely accepted mechanism (Scheme 4), the first reaction step consists of the 1,4-addition of the catalytic tertiary amine (DABCO) (a) to the activated alkene (methyl acrylate) (b), which generates a zwitterionic intermediate (c). The next step involves the aldolic addition of zwitterion and aldehyde (d) to yield an intermediate (e). Later, the intermediate (e) undergoes an intramolecular prototropic shift to form another intermediate (e′), which is isomeric to (e). In the last step, the intermediate (e′) forms the final MBH adduct (f) by releasing the intact base back into the solution.
 |
| | Scheme 4 Proposed reaction pathway for the MBH reaction of aldehyde 2 with methyl acrylate in the presence of DABCO. | |
For ESI-MS experiments, aldehyde 1 or 2 (1 equivalent), methyl acrylate (8 equivalents) and DABCO (1 equivalent) were mixed without additional solvent and allowed to react. The positive ion ESI mass spectra were obtained for the reaction mixtures at different reaction times (0.25, 3, 6, 9, 12 and 24 hours for aldehyde 2, and 1, 3, 6, 9, 12, 24 and 48 hours for aldehyde 1). The mass spectra included the peaks corresponding to the starting materials, i.e., the [M + H]+ ion of DABCO (a) at m/z 113 and the [M + Na]+ ion of dibenzofuran aldehyde (d) at m/z 219. The peak corresponding to methyl acrylate (b) could not be seen in the spectra, possibly because it was not amenable to ionization under positive ion ESI conditions. The peak due to the expected product was observed at m/z 305 (f), which corresponds to the [M + Na]+ ion. The intermediate species appeared at m/z 199 (c) and 395 (e or e′) (Fig. 4).
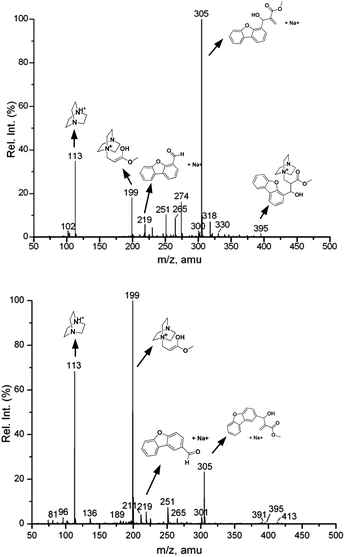 |
| | Fig. 4 Positive ion ESI mass spectra of the reaction mixture of aldehyde 2 (top) and aldehyde 1 (bottom) after 3 hours. | |
Though both the isomeric dibenzofuran aldehydes 1 and 2 showed similar sets of ions, consistent differences were found in the ion yields of the reaction intermediates/products, i.e., the ions appearing at m/z 199 (c), 305 (f) and 395 (e). The ion yields of these ions were plotted against the reaction time point of the two reactions (Fig. 5 and 6) (Table 2).
 |
| | Fig. 5 Ion counts at different time intervals for the reaction with aldehyde 2. | |
 |
| | Fig. 6 Ion counts at different time intervals for the reaction with aldehyde 1. | |
Table 2 ESI-HRMS data of the ions detected in the reaction of aldehyde 2 at 3 hours (similar data was obtained for aldehyde 1; data not provided)
| Observed m/z |
Ion |
Chemical formula |
Theoretical m/z |
Error (in ppm) |
| 113.1080 |
[M + H]+ |
C6H13N2 |
113.1078 |
1.1 |
| 199.1456 |
[M + H]+ |
C10H19N2O2 |
199.1446 |
4.8 |
| 219.0418 |
[M + Na]+ |
C13H8O2Na |
219.0421 |
−1.8 |
| 305.0801 |
[M + Na]+ |
C17H14O4Na |
305.0789 |
3.7 |
| 395.1990 |
[M + H]+ |
C23H27N2O4 |
395.1971 |
4.9 |
As expected, the ion yields of m/z 305 significantly increased with reaction time in aldehyde 2, and the same is true for the ion at m/z 199. The intermediate ion at m/z 395, which is expected to be crucial, was found to be in low abundance in both the aldehydes; therefore, the changes in the ion yields are not prominent.
Thus, we moved to collision induced dissociation (CID) experiments on the ions at m/z 395 obtained from both the reactions to understand their stabilities. The spectra were obtained at different collision energy values (5–15 eV). The CID spectra of the ions m/z 395 from both the reactions showed exclusively one product ion at m/z 113 (Fig. 7). The percentage of total ion current (%TIC) of m/z 395 was calculated from the ion abundances of m/z 113 and 395, and this value is plotted against the collision energy (Fig. 8). Fig. 8 clearly reveals that the intermediate ion from aldehyde 2 is relatively more stable than that from aldehyde 1. However, more than one transition state (TS) is expected to appear at the m/z 395; hence, it is difficult to pinpoint which transition state is crucial for the differential reactivity of aldehydes 1 and 2. Hence, we moved to theoretical studies for better understanding of the BH reaction mechanism.
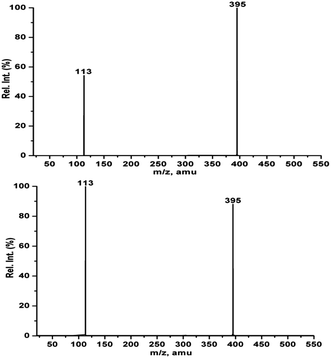 |
| | Fig. 7 CID mass spectra of m/z 395 from aldehyde 2 (top) and aldehyde 1 (bottom) obtained at the collision energy of 5 eV. | |
 |
| | Fig. 8 Plot of percentage of total ion current (%TIC) values vs. collision energy from the MS/MS spectra of the intermediates at m/z 395 from aldehyde 1 and aldehyde 2. | |
Computational studies
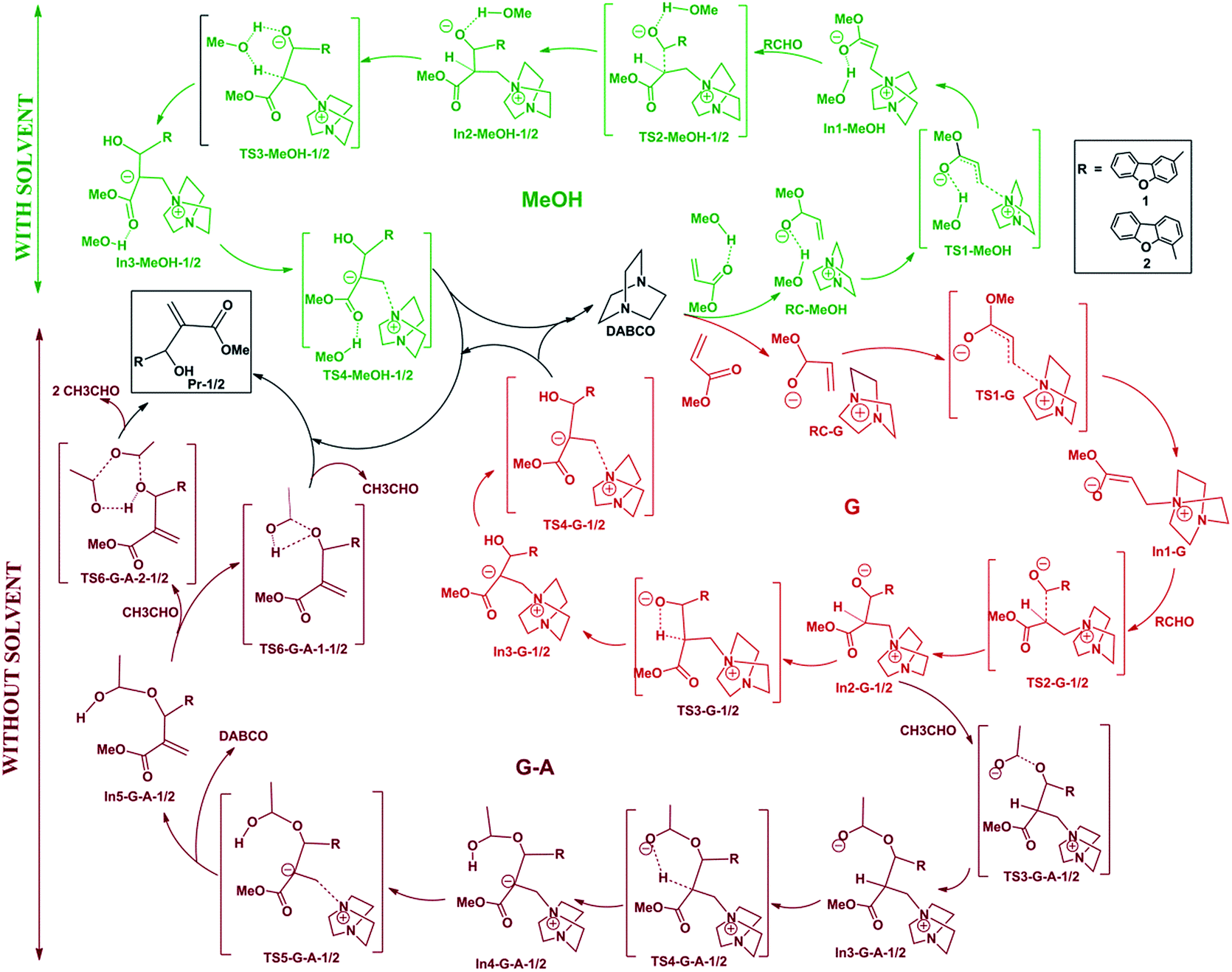
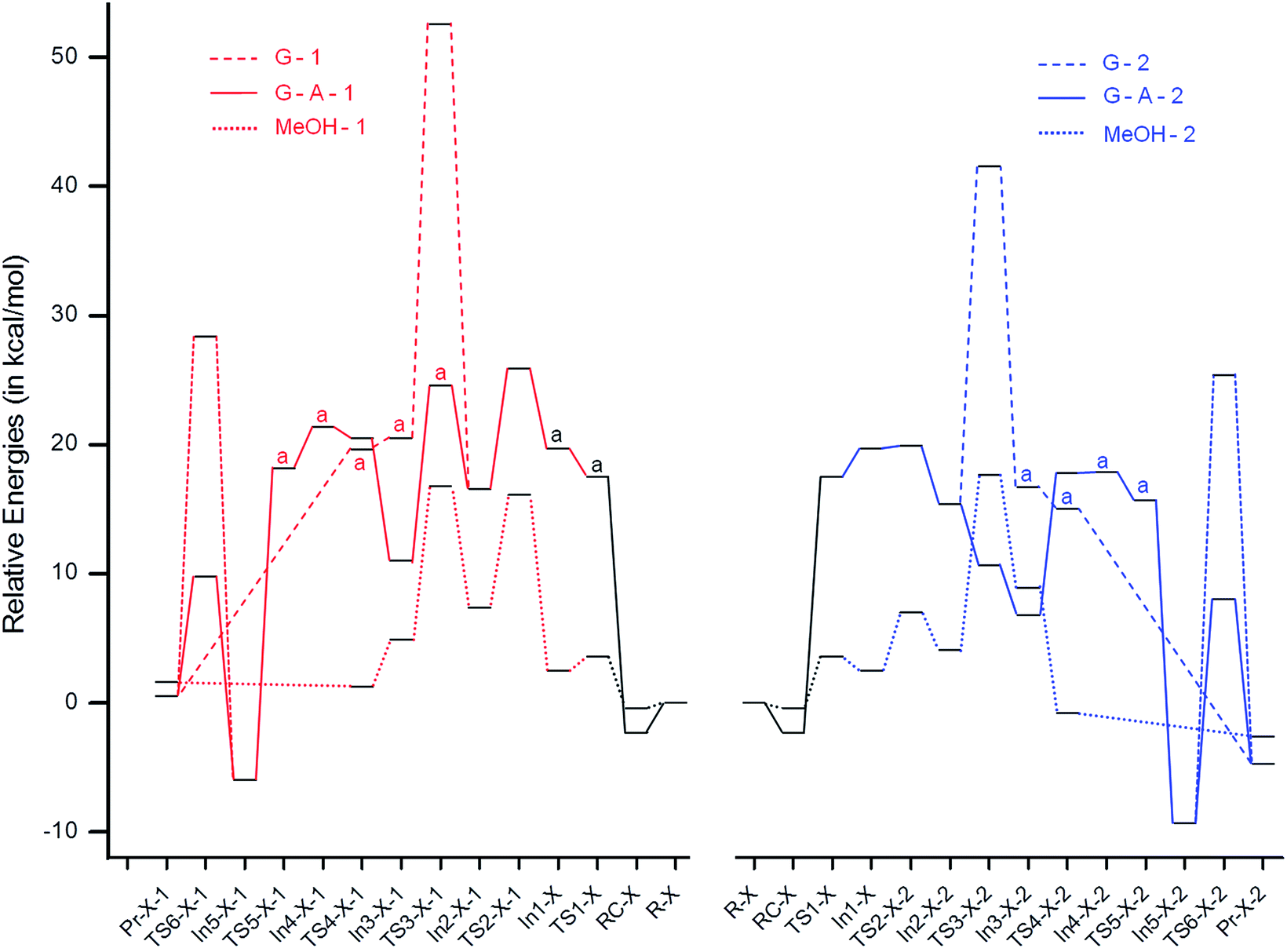
To gain insights into the observed differences in the rates of formation of isomeric MBH adducts, quantum chemical calculations have been performed, considering different mechanisms using both the gas phase and the solvent phase based on the previously reported pathways16 (Scheme 5). The nomenclature (–X–) is given as ‘–MeOH–’ for the structures obtained using methanol (as both explicit and implicit solvent), ‘–G–’ for the structures obtained using the gas phase and ‘–G–A–’ for those obtained using the gas phase and a second aldehyde molecule, which is reported to play a crucial role in the proton transfer step.16a,b The structures bearing aldehydes 1 and 2 are named with the suffixes ‘-1’ and ‘-2’, respectively. IRC calculations have been performed to validate the transition states. The reaction profiles generated are depicted in Fig. 9.
 |
| | Scheme 5 Gas and solvent (methanol) phase reaction paths considered for the computational study. | |
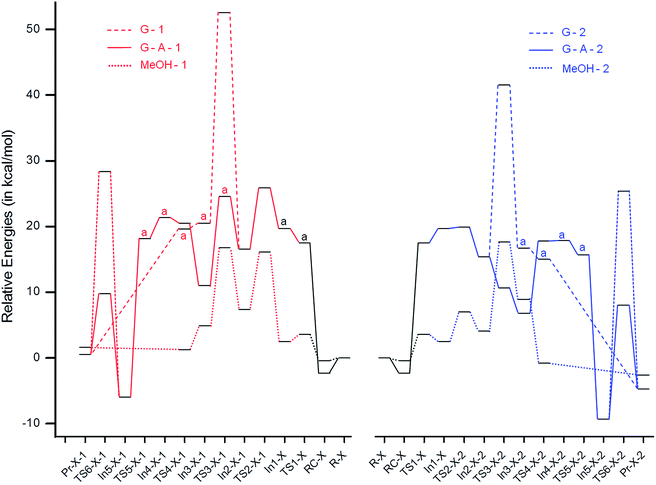 |
| | Fig. 9 Gas and solvent phase reaction energy profiles for the considered MBH reaction obtained using aldehydes 1 (red) and 2 (blue) at the mPW1K/6-31 + G(d,p) level of theory. A represents the structures obtained at the mPW1K/6-31 + g(d,p)//HF/6-31 + G(d,p) level of theory. | |
The gas and solvent phase reaction profiles were initially generated considering the paths ‘G’ and ‘MeOH’, as depicted in Scheme 5. After the formation of In1-X from methyl acrylate (MA) and DABCO, the aldehyde 1/2 attacks In1-X, proceeding through a C–C bond formation step, and forms In2-X-1/In2-X-2 (−3.1/−4.2 and 4.9/1.6 kcal mol−1 for G and MeOH, respectively) via transition state TS2-X-1/TS2-X-2 (6.2/0.3 and 13.7/4.5 kcal mol−1 for G and MeOH, respectively). In2-X-1/In2-X-2 then undergoes proton migration and forms In3-X-1/In3-X-2 (3.9/1.3 and −2.5/4.8 kcal mol−1 for G and MeOH, respectively) via transition state TS3-X-1/TS3-X-2 (36.0/26.2 and 9.4/13.6 kcal mol−1 for G and MeOH, respectively). These results clearly show that the proton migration step is the rate determining step (RDS) in both the gas and the solvent phases. However, the activation energy for the C–C bond formation TS for aldehyde 1 in the solvent phase is ∼4 kcal mol−1 higher in energy than the proton migration TS, suggesting that both TSs play a key role during the reaction process in this case. The higher energy for the proton migration TS in the gas phase compared to the solvent phase might be due to the four membered transition state in the former case. In3-X-1/In3-X-2 after DABCO elimination forms the final MBH adduct. The thermodynamic and kinetic energies, in general, along the reaction profile are observed to be much lower for 2 compared to 1, suggesting that the reaction with 2 should be faster than that with 1. These are in accordance with the experimental results where 2 reacts faster to yield the final MBH adduct compared to 1. Although the computational results suggest a huge effect of solvent on the considered MBH reaction, the experimental results suggest no effect of solvent on the reaction. Hence, we proceeded to look further at an alternative mechanism,16a,b considering a second equivalent of aldehyde. Previous reports by McQuade and coworkers16a and Harvey and coworkers16b suggest the role played by the second equivalent of aldehyde in the proton transfer step. Considering the bulkiness of the aldehyde, the aromatic moiety (R) is replaced with a methyl group, which will reduce the computational cost.
The initial step before the proton transfer step is the addition of a second aldehyde (A) to In2-G, resulting in In3-G-A-1/In3-G-A-2 (−5.6/−8.6 kcal mol−1) via transition state TS3-G-A-1/TS3-G-A-2 (8.0/−4.7 kcal mol−1). In3-G-A-1/In3-G-A-2 then undergoes proton migration and forms In4-G-A-1/In4-G-A-2 (10.4/11.1 kcal mol−1) via a 6-membered transition state, TS4-G-A-1/TS4-G-A-2 (9.5/11.0 kcal mol−1). Similar to the results reported by Harvey and coworkers16b in the absence of protic species, the activation energy for the 4-membered proton transfer transition state, TS3-G-1/TS3-G-2, is much higher in energy compared to the 6-membered transition state TS4-G-A-1/TS4-G-A-2. In4-G-A-1/In4-G-A-2 undergoes DABCO elimination with a subsequent proton transfer and aldehyde elimination and forms the final MBH adduct. The transition state involving the elimination of the second aldehyde and the simultaneous proton transfer for TS6-G-A-1-1/TS6-G-A-1-2 is 34.4/34.7 kcal mol−1, which is much higher (∼18.0 kcal mol−1) in energy than TS4-G-A-1/TS4-G-A-2. This step cannot be the RDS, since previous kinetic studies16a report the proton transfer step to be the RDS; therefore, an alternative TS involving two molecules of aldehyde, thereby resulting in a 6-membered TS, was looked at. The TS involving an additional molecule of aldehyde lowers the energy drastically (by ∼15 kcal mol−1) for TS6-G-A-2-1 and TS6-G-A-2-2, with activation energy barriers of 15.7 and 17.3 kcal mol−1, respectively. The gas phase results are thus in accordance with McQuade's proposal of MBH mechanism, wherein in the absence of protic solvent, the second equivalent of aldehyde plays a key role in the proton transfer step. The virtually similar energy gaps for the G–A and MeOH paths might be the reason for the similar yields observed experimentally both in the presence and absence of protic solvent.
Similar to the G and MeOH energetics, the thermodynamic and kinetic energies along the reaction profile for G–A are observed to be much lower for 2 compared to 1, suggesting that the reaction with 2 should be faster than with 1. To understand the reason for the lower energies of In2-X-2 and TS2-X-2 compared to In2-X-1 and TS2-X-1, respectively, bond critical points for TS2-X-1, In2-X-1, TS2-X-2 and In2-X-2 were looked at for both the gas and solvent phases. The electron densities and their Laplacian values at the bond critical points (BCPs) are depicted in Fig. 10 (gas phase) and Fig. S1† (solvent phase). The lower energies of aldehyde 2 for both the gas and solvent phases might be due to the interactions of the endocyclic oxygen (OEC), which are observed in case of 2, wherein aldehyde 1 is devoid of such interactions. This in turn might play a role in hastening the reaction process for 2.
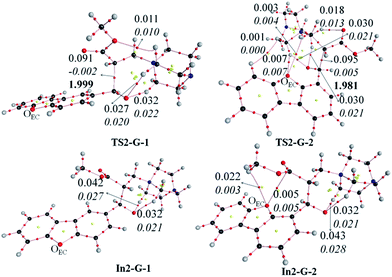 |
| | Fig. 10 Bond critical points for TS2-G-1, TS2-G-2, In2-G-1 and In2-G-2. Electron density values: normal, Laplacian of electron density: italics, bond lengths: bold. | |
Conclusions
In summary, we herein report the Morita–Baylis–Hillman reactions of two isomeric dibenzofuran carbaldehydes (1 and 2) with different electrophiles in the presence of DABCO as a base catalyst. HPLC analysis of the reaction mixtures revealed that aldehyde 2 reacted at a faster rate compared to its isomeric aldehyde 1 to give the respective MBH adducts. In situ ESI-MS and MS/MS experiments concluded that the change in the rate of reaction was due to the stability of an intermediate (ion at m/z 395) formed during the course of the MBH reaction. The gas and solvent phase reaction profiles gave insights into the observed differences in the rates of formation of isomeric MBH adducts, suggesting aldehyde 2 to be more reactive than aldehyde 1. The proton migration step was found to be the rate determining step. In the absence of protic species, an additional aldehyde molecule plays a key role in the proton migration, which is in accordance with McQuade's proposal of the MBH mechanism.
Experimental section
General remarks
Melting points were measured with a Fischer-Johns melting point apparatus and are uncorrected. IR spectra were obtained as KBr pellets and absorptions are reported in cm−1. NMR spectra were obtained on 300 MHz (Bruker) and 500 MHz (Varian) spectrometers in appropriate solvents using TMS as the internal standard; the chemical shifts are shown in δ scales. 13C NMR spectra were obtained on 75 MHz spectrometers. High-resolution mass spectra were obtained using ESI-QTOF mass spectrometry. All the experiments were monitored by analytical thin layer chromatography (TLC) performed on silica gel GF254 pre-coated plates. After elution, the plate was visualized under UV illumination at 254 nm for UV active materials. Silica gel finer than 200 mesh was used for column chromatography. Yields refer to chromatographically and spectroscopically homogeneous materials unless otherwise stated. Appropriate names for all the new compounds are given with the help of ChemBioOffice 12.0, 2010. The solvents for column elution were purchased commercially, and for mass spectral studies, the solvents were purchased from Sigma Aldrich. Aldehydes 1 and 2 were synthesized by following the reported protocols.13,14
Mass spectrometry experiments
All the mass spectrometry experiments were performed using a quadrupole time-of-flight mass spectrometer (QSTAR XL, Applied Biosystems/MDS Sciex, Foster City, CA, USA) equipped with an ESI ion source. The data acquisition was under the control of Analyst QS software (Applied Biosystems). All the samples were introduced into the source by flow injection (10 μL loop) using methanol as the mobile phase at a flow rate of 30 μL min−1. The typical source conditions were capillary voltage, 5 kV; declustering potentials DP1, 60 V; DP2, 10 V; focusing potential, 250 V; and mass resolution 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (FWHM). Nitrogen was used as the curtain gas and the collision gas. For the CID experiments, the precursor ion of interest was selected using the quadrupole analyser and the product ions were analysed using the TOF analyser. The collision energies used were between 5 and 15 eV.
000 (FWHM). Nitrogen was used as the curtain gas and the collision gas. For the CID experiments, the precursor ion of interest was selected using the quadrupole analyser and the product ions were analysed using the TOF analyser. The collision energies used were between 5 and 15 eV.
Procedure for computational studies
Calculations were performed on all the systems at the mPW1K/6-31 + G(d,p) level of theory, as this method has been proved to be better in locating zwitterionic structures on a potential energy surface.17 The TSs and Ins that could not be optimized at the aforementioned level were obtained at the mPW1K/6-31 + g(d,p)//HF/6-31 + G(d,p) level of theory. Solvent phase calculations were performed using PCM. All the calculations were performed using the Gaussian 09 programme package.18 The BCPs were generated using AIM2000 software.19
General procedure for the preparation of MBH adducts 1A-D and 2A-D
A solution of aldehyde 1 or 2 (5 mmol), DABCO (5 mmol) and corresponding activated olefin (40 mmol) was stirred at RT for the required time (Scheme 2). After that, the reaction mixture was diluted with ethyl acetate (20 mL) and washed successively with 2 N HCl (2 × 5 mL), water (2 × 5 mL) and saturated NaHCO3 solution (5 mL); the combined organic layers were dried over anhydrous Na2SO4. The solvent was removed under vacuum and the crude residue thus obtained was chromatographed using EtOAc and hexane (10:90) over silica gel to give the corresponding MBH adducts 1A-D and 2A-D in very good yields.
Methyl 2-(dibenzo[b,d]furan-2-yl(hydroxy)methyl)acrylate (1A)
Yield 75%; light yellow syrup. 1H NMR (300 MHz, CDCl3) δ 7.94–7.82 (m, 2H), 7.58–7.44 (m, 2H), 7.43–7.35 (m, 2H), 7.28 (t, J = 7.5 Hz, 1H), 6.31 (s, 1H), 5.86–5.82 (m, 1H), 5.65 (s, 1H), 3.70 (s, 3H), 3.10 (br s, 1H). 13C NMR (75 MHz, CDCl3) δ 166.7, 156.5, 142.1, 140.9, 135.8, 127.2, 126.0, 125.8, 122 0.7, 121.2, 120.7, 118.8, 111.6, 111.4, 109.2, 73.1, 51.9. IR (neat) 3450, 2951, 1718, 1629, 1478, 1446, 1196, 1149, 1040, 816, 750 cm−1. MS (ESI) m/z 305 [M + Na]+; HRMS (ESI) calcd for C17H14O4Na: 305.0789, found: 305.0786.
Ethyl 2-(dibenzo[b,d]furan-2-yl(hydroxy)methyl)acrylate (1B)
Yield 70%; white solid; mp 76–78 °C; 1H NMR (300 MHz, CDCl3) δ 7.99–7.84 (m, 2H), 7.61–7.49 (m, 2H), 7.47–7.37 (m, 2H), 7.35–7.27 (m, 1H), 6.35 (s, 1H), 5.85 (s, 1H), 5.73–5.67 (br s, 1H), 4.17 (q, J = 7.5 & 6.7 Hz, 2H), 3.15 (br, 1H), 1.25 (t, J = 7.5 & 6.7 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 165.9, 156.3, 155.4, 142.5, 136.0, 126.9, 125.8, 125.0, 123.8, 122.4, 121.3, 120.4, 118.8, 111.4, 111.1, 72.5, 60.6, 13.8. IR (KBr) 3444, 2983, 1714, 1633, 1449, 1198, 1022, 960 cm−1. MS (EI) m/z 296 [M]+; HRMS (EI) calcd for C18H16O4: 296.10486, found: 296.10536.
2-(Dibenzo[b,d]furan-2-yl(hydroxy)methyl)acrylonitrile (1C)
Yield 93%; white solid; mp 84–86 °C; 1H NMR (300 MHz, CDCl3) δ 7.90 (d, J = 10.5 Hz, 2H), 7.56–7.50 (m, 2H), 7.48–7.38 (m, 2H), 7.31 (t, J = 7.5 Hz, 1H), 6.11 (s, 1H), 6.01 (s, 1H), 5.39 (s, 1H), 2.80 (br s, 1H). 13C NMR (75 MHz, CDCl3) δ 156.4, 156.0, 133.7, 129.8, 127.4, 126.2, 125.5, 124.5, 123.6, 122.8, 120.7, 118.8, 116.9, 111.9, 111.6, 74.0. IR (KBr) 3469, 2234, 1902, 1601, 1476, 1446, 1432, 1246, 1201, 1055, 952, 815, 744 cm−1. MS (EI) m/z 249 [M]+; HRMS (EI) calcd for C16H11NO2: 248.07898, found: 248.07890.
2-(Dibenzo[b,d]furan-2-yl(hydroxy)methyl)cyclohex-2-enone (1D)
Yield 63%; syrup. 1H NMR (300 MHz, CDCl3) δ 8.02–7.87 (m, 2H), 7.65–7.28 (m, 5H), 6.71 (s, 1H), 5.72 (s, 1H), 2.52–2.37 (m, 4H), 2.10–1.99 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 200.2, 156.3, 155.3, 147.3, 136.2, 127.0, 125.6, 122.5, 121.1, 120.6, 118.6, 115.9, 111.4, 109.5, 71.8, 38.3, 25.5, 22.3. IR (neat) 3411, 2924, 2853, 1666, 1510, 1478, 1444, 1374, 1244, 1196, 1020, 841, 750 cm−1. MS (EI) m/z 292 [M]+; HRMS (EI) calcd for C19H16O3: 292.10994, found: 292.10980.
Methyl 2-(dibenzo[b,d]furan-4-yl(hydroxy)methyl)acrylate (2A)
Yield 99%; white solid; mp 95–97 °C; 1H NMR (300 MHz, CDCl3) δ 7.90 (d, J = 7.5 Hz, 1H), 7.84 (dd, J = 1.5 & 7.5 Hz, 1H), 7.52 (t, J = 8.3 Hz, 2H), 7.41 (td, J = 1.5 & 6.7 Hz, 1H), 7.36–7.25 (m, 2H), 6.32 (s, 1H), 6.17–6.12 (m, 1H), 5.80 (s, 1H), 3.75 (s, 3H), 3.44 (d, J = 6.0 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ 166.4, 155.8, 153.1, 140.7, 136.8, 126.1, 125.2, 124.9, 123.9, 123.9, 122.6, 122.5, 120.4, 119.8, 111.5, 67.6, 51.6. IR (KBr) 3440, 3059, 2952, 1717, 1633, 1589, 1451, 1268, 1150, 1107, 1036, 960, 846, 756 cm−1. MS (ESI) m/z 305 [M + Na]+; HRMS (ESI) calcd for C17H14O4Na: 305.0789, found: 305.0783.
Ethyl 2-(dibenzo[b,d]furan-4-yl(hydroxy)methyl)acrylate (2B)
Yield 97%; light red syrup. 1H NMR (300 MHz, CDCl3) δ 7.88 (d, J = 7.7 Hz, 1H), 7.82 (d, J = 7.7 Hz, 1H), 7.55–7.47 (m, 2H), 7.40 (t, J = 6.7 Hz, 1H), 7.33–7.26 (m, 2H), 6.31 (s, 1H), 6.14 (s, 1H), 5.79 (s, 1H), 4.17 (q, J = 6.7 Hz, 2H), 3.48 (br, 1H), 1.22 (t, J = 6.7 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 166.1, 155.8, 153.1, 140.9, 126.9, 126.0, 125.2, 124.9, 124.0, 123.9, 122.6, 122.6, 122.5, 120.4, 119.9, 111.5, 67.8, 60.7, 13.7. IR (neat) 3504, 3062, 2989, 2912, 1706, 1628, 1423, 1283, 1184, 1035, 960, 834, 749 cm−1. MS (EI) m/z 296 [M]+; HRMS (EI) calcd for C18H16O4: 296.10486, found: 296.10519.
2-(Dibenzo[b,d]furan-4-yl(hydroxy)methyl)acrylonitrile (2C)
Yield 97%; colourless syrup. 1H NMR (300 MHz, CDCl3) δ 7.94–7.85 (m, 2H), 7.56–7.49 (m, 2H), 7.43 (td, J = 7.3 & 1.3 Hz, 1H), 7.39–7.28 (m, 2H), 6.14 (d, J = 1.1 Hz, 1H), 6.03 (d, J = 0.9 Hz, 1H), 5.88 (s, 1H), 3.12 (br s, 1H). 13C NMR (75 MHz, CDCl3) δ 155.9, 152.9, 130.6, 127.4, 125.0, 124.5, 123.7, 123.2, 123.0, 121.0, 120.7, 116.8, 111.6, 69.3. IR (neat) 3434, 2925, 2228, 1716, 1584, 1451, 1425, 1265, 1150, 1044, 951, 837, 754 cm−1. MS (EI) m/z 249 [M]+; HRMS (EI) calcd for C16H11NO2: 248.07898, found: 248.07890.
2-(Dibenzo[b,d]furan-4-yl(hydroxy)methyl)cyclohex-2-none (2D)
Yield 80%; black solid; mp 66–68 °C; 1H NMR (300 MHz, CDCl3) δ 7.91 (d, J = 7.5 Hz, 1H), 7.84 (d, J = 7.5 Hz, 1H), 7.61–7.48 (m, 2H), 7.46–7.26 (m, 3H), 6.65 (t, J = 4.5 Hz, 1H), 6.12 (s, 1H), 3.83 (br s, 1H), 2.53–2.43 (m, 2H), 2.35 (q, J = 5.2 Hz, 2H), 2.08–1.98 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 199.9, 155.8, 152.8, 147.1, 139.6, 126.8, 125.5, 125.0, 124.0, 123.8, 122.6, 122.5, 120.4, 119.5, 111.5, 67.3, 38.2, 25.5, 22.3. IR (KBr) 3418, 3059, 2926, 1667, 1588, 1474, 1423, 1378, 1250, 1186, 1024, 845, 755 cm−1. MS (EI) m/z 292 [M]+; HRMS (EI) calcd for C19H16O3: 292.10994, found: 292.10980.
Acknowledgements
Authors thank Dr G. Narahari Sastry for his critical reading and support in computational study. Financial assistance for 12th FYP projects (ORIGIN, CSC0108 and AARF, CSC0406) from CSIR, New Delhi, India is gratefully acknowledged. TY and VD are thankful to CSIR, New Delhi, India for senior research fellowships.
Notes and references
-
(a) A. B. Baylis and M. E. D. Hillman, German patent 2155113, 1972, Chem. Abstr., 1972, vol. 77, p. 34174q;
(b) V. Singh and S. Batra, Tetrahedron, 2008, 64, 4511–4574 CrossRef CAS;
(c) Y. Wei and M. Shi, Chem. Rev., 2013, 113, 6659–6690 CrossRef CAS PubMed;
(d) D. Basavaiah and G. Veeraraghavaiah, Chem. Soc. Rev., 2012, 41, 68–78 RSC;
(e) D. Basavaiah, B. S. Reddy and S. S. Badsara, Chem. Rev., 2010, 110, 5447–5674 CrossRef CAS PubMed.
-
(a) D. Basavaiah, A. J. Rao and T. Satyanarayana, Chem. Rev., 2003, 103, 811–892 CrossRef CAS PubMed;
(b) W. P. Almeida and F. Coelho, Quim. Nova, 2000, 23, 98–101 (Chem. Abstr., 2000, 132, 236562e) CrossRef;
(c) E. Ciganek, Org. React., 1997, 51, 201–350 CAS;
(d) D. Basavaiah, P. D. Rao and R. S. Hyma, Tetrahedron, 1996, 52, 8001–8062 CrossRef CAS.
-
(a) K. E. Price, S. J. Broadwater, B. J. Walker and D. T. McQuade, J. Org. Chem., 2005, 70, 3980–3987 CrossRef CAS;
(b) S. Luo, P. G. Wang and J.-P. Cheng, J. Org. Chem., 2004, 69, 555–558 CrossRef CAS PubMed;
(c) Y. Iwabuchi, M. Nakatani, N. Yokoyama and S. Hatakeyama, J. Am. Chem. Soc., 1999, 121, 10219–10220 CrossRef CAS;
(d) V. K. Aggarwal, A. Mereu, G. J. Tarver and R. McCague, J. Org. Chem., 1998, 63, 7183–7189 CrossRef CAS PubMed;
(e) S. Rafel and J. W. Leahy, J. Org. Chem., 1997, 62, 1521–1522 CrossRef CAS;
(f) R. O. M. A. de Souza and M. L. A. A. Vasconcellos, Synth. Commun., 2003, 33, 1383–1389 CrossRef.
-
(a) F. Coelho, W. P. Almeida, D. Veronese, C. R. Mateus, E. C. S. Lopes, R. C. Rossi, G. P. C. Silveira and C. H. Pavam, Tetrahedron, 2002, 58, 7437–7447 CrossRef CAS;
(b) W. P. Almeida and F. Coelho, Tetrahedron Lett., 1998, 39, 8609–8612 CrossRef CAS.
- T. Cablewski, A. F. Faux and C. R. Strauss, J. Org. Chem., 1994, 59, 3408–3412 CrossRef CAS.
-
(a) A. Kumar and S. S. Pawar, Tetrahedron, 2003, 59, 5019–5026 CrossRef CAS;
(b) M. R. Netherton and G. C. Fu, Org. Lett., 2001, 3, 4295–4298 CrossRef CAS PubMed;
(c) M. Shi and Y. S. Feng, J. Org. Chem., 2001, 66, 406–411 CrossRef CAS PubMed.
-
(a) L. S. Santos, B. A. da SilveiraNeto, C. S. Consorti, C. H. Pavam, W. P. Almeida, F. Coelho, J. Dupont and M. N. Eberlin, J. Phys. Org. Chem., 2006, 19, 731–736 CrossRef CAS;
(b) R. O. M. A. de Souza, P. H. Fregadolli, K. M. Goncalves, L. C. Sequeira, V. L. P. Pereira, L. C. Filho, P. M. Esteves, M. L. A. A. Vasconcellos and O. A. C. Antunes, Lett. Org. Chem., 2006, 3, 936–939 CrossRef CAS;
(c) S. H. Zhao, H. R. Zhang, L. H. Feng and Z. B. Chen, J. Mol. Catal. A: Chem., 2006, 258, 251–256 CrossRef CAS;
(d) H. Gong, C. Q. Cai, N. F. Yang, L. W. Yang, J. Zhang and Q. H. Fan, J. Mol. Catal. A: Chem., 2006, 249, 236–239 CrossRef CAS;
(e) X. Mi, S. Luo, H. Xu, L. Zhang and J. P. Cheng, Tetrahedron, 2006, 62, 2537–2544 CrossRef CAS;
(f) V. K. Aggarwal, I. Emme and A. Mereu, Chem. Commun., 2002, 1612–1613 RSC;
(g) R. S. Porto, G. W. Amarante, M. Cavallaro, R. J. Poppi and F. Coelho, Tetrahedron Lett., 2009, 50, 1184–1187 CrossRef CAS.
-
(a) Y. Wei and M. Shi, Chem. Rev., 2013, 113, 6659–6690 CrossRef CAS PubMed;
(b) K. Matsui, S. Takizawa and H. Sasai, J. Am. Chem. Soc., 2005, 127, 3680–3681 CrossRef CAS PubMed;
(c) Y. Wei and M. Shi, Acc. Chem. Res., 2010, 43, 1005–1018 CrossRef CAS PubMed;
(d) M. Shi, G. N. Ma and J. Gao, J. Org. Chem., 2007, 72, 9779–9981 CrossRef CAS PubMed;
(e) A. Berkessel, K. Roland and J. M. Neudörfl, Org. Lett., 2006, 8, 4195–4198 CrossRef CAS PubMed;
(f) Y.-L. Shi and M. Shi, Adv. Synth. Catal., 2007, 349, 2129–2135 CrossRef CAS;
(g) J. Wang, H. Li, X. Yu, L. Zu and W. Wang, Org. Lett., 2005, 7, 4293–4296 CrossRef CAS PubMed;
(h) G. Masson, C. Housseman and J. Zhu, Angew. Chem., Int. Ed. Engl., 2007, 46, 4614–4628 CrossRef CAS PubMed;
(i) Y.-H. Liu and M. Shi, Adv. Synth. Catal., 2008, 350, 122–128 CrossRef CAS;
(j) C. K.-W. Kwong, R. Huang, M. Zhang, M. Shi and P. H. Toy, Chem.–Eur. J., 2007, 13, 2369–2376 CrossRef CAS PubMed;
(k) J.-M. Garnier, C. Anstiss and F. Liu, Adv. Synth. Catal., 2009, 351, 331–338 CrossRef CAS.
-
(a) L. S. Santos, C. H. Pavam, W. P. Almeida, F. Coelho and M. N. Eberlin, Angew. Chem., Int. Ed., 2004, 43, 4330–4333 CrossRef CAS PubMed;
(b) K. E. Price, S. J. Broadwater, H. M. Jung and D. T. McQuade, Org. Lett., 2005, 7, 147–150 CrossRef CAS PubMed;
(c) K. E. Price, S. J. Broadwater, B. J. Walker and D. T. McQuade, J. Org. Chem., 2005, 70, 3980–3987 CrossRef CAS PubMed;
(d) V. K. Aggarwal, S. Y. Fulford and G. C. Lloyd-Jones, Angew. Chem., Int. Ed., 2005, 44, 1706–1708 CrossRef CAS PubMed;
(e) G. W. Amarante, M. Benassi, H. M. Milagre, A. A. Braga, F. Maseras, M. N. Eberlin and F. Coelho, Chem.–Eur. J., 2009, 15, 12460–12469 CrossRef CAS PubMed;
(f) D. Roy and R. B. Sunoj, Chem.–Eur. J., 2008, 14, 10530–10534 CrossRef CAS PubMed;
(g) D. Roy, C. Patel and R. B. Sunoj, J. Org. Chem., 2009, 74, 6936–6943 CrossRef CAS PubMed;
(h) L. Dong, S. Qin, Z. Su, H. Yang and C. Hu, Org. Biomol. Chem., 2010, 8, 3985–3991 RSC;
(i) K. E. Price, S. J. Broadwater, B. J. Walker and D. T. McQuade, J. Org. Chem., 2005, 70, 3980–3987 CrossRef CAS PubMed;
(j) D. Cantillo and C. O. Kappe, J. Org. Chem., 2010, 75, 8615–8626 CrossRef CAS;
(k) G. W. Amarante, H. M. Milagre, B. G. Vaz, V. B. R. Ferreira, M. N. Eberlin and F. Coelho, J. Org. Chem., 2009, 74, 3031–3037 CrossRef CAS PubMed.
- P. Narender, B. Gangadasu, U. Srinivas, M. Ravinder, S. B. Kumar, C. Ramesh, V. J. Rao and J. M. Rao, U.S. Patent, 7, 666,883, 2010.
- A. Patra, S. Batra, B. Kundu, B. S. Joshi, R. Roy and A. P. Bhaduria, Synthesis, 2001, 2, 276–280 CrossRef.
- S. Nag, V. Singh and S. Batra, ARKIVOC, 2007, xiv, 185–203 Search PubMed.
-
(a) T. Yempala, J. P. Sridevi, P. Yogeeswari, D. Sriram and S. Kantevari, Eur. J. Med. Chem., 2014, 71, 160–167 CrossRef CAS;
(b) S. Kantevari, T. Yempala, G. Surineni, B. Sridhar, P. Yogeeswari and D. Sriram, Eur. J. Med. Chem., 2011, 46, 4827–4833 CrossRef CAS PubMed;
(c) T. Yempala, D. Sriram, P. Yogeeswari and S. Kantevari, Bioorg. Med. Chem. Lett., 2012, 22, 7426–7430 CrossRef CAS PubMed.
- N. Lakshminarayana, Y. R. Prasad, L. Gharat, A. Thomas, S. Narayanana, A. Raghurama, C. V. Srinivasan and B. Gopalan, Eur. J. Med. Chem., 2010, 45, 3709–3918 CrossRef CAS PubMed.
-
(a) C. S. Ho, C. W. K. Lam, M. H. M. Chan, R. C. K. Cheung, L. K. Law, L. C. W. Lit, K. F. Ng, M. W. M. Suen and H. L. Tai, Clin. Biochem. Rev., 2003, 24, 3–12 CAS;
(b) J. B. Fenn, M. Mann, C. K. Meng, S. F. Wong and C. M. Whitehouse, Science, 1989, 246, 64–71 CAS;
(c) R. B. Cole, Electrospray Ionization Mass Spectroscopy, Wiley, New York, 1997 Search PubMed;
(d) R. G. Cooks, D. X. Zhang, K. J. Koch, F. C. Gozzo and M. N. Eberlin, Anal. Chem., 2001, 73, 3646–3655 CrossRef CAS;
(e) Z. Takats, S. C. Nanita and R. G. Cooks, Angew. Chem., Int. Ed., 2003, 42, 3521–3523 CrossRef CAS.
-
(a) K. E. Price, S. J. Broadwater, H. M. Jung and D. T. McQuade, Org. Lett., 2005, 7, 147–150 CrossRef CAS PubMed;
(b) R. Robiette, V. K. Aggarwal and J. N. Harvey, J. Am. Chem. Soc., 2007, 129, 15513–15525 CrossRef CAS;
(c) V. K. Aggarwal, S. Y. Fulford and G. C. Lloyd-Jones, Angew. Chem., Int. Ed., 2005, 44, 1706–1708 CrossRef CAS PubMed.
-
(a) Y. Wei, B. Sateesh, B. Maryasin, G. N. Sastry and H. Zipse, J. Comput. Chem., 2009, 30, 2617–2624 CrossRef CAS PubMed;
(b) Y. Wei, T. Singer, H. Mayr, G. N. Sastry and H. Zipse, J. Comput. Chem., 2008, 29, 291–297 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- AIM 2000 Version 2.0, F. Biegler-König, J. Schönbohm, University of Applied Science, Bielefeld, Germany, 2002 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H, 13C NMR and mass (ESI-MS and HRMS) spectra of all the new compounds were incorporated in that. See DOI: 10.1039/c5ra14486h |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.