
Coro-graphene and circumcoro-graphyne: novel two-dimensional materials with exciting electronic properties†
Abstract
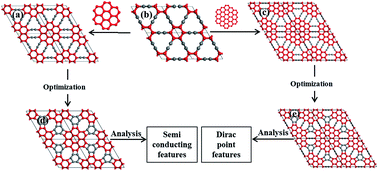
In this study, two novel forms of two-dimensional (2D) carbon frameworks (named as coro-graphene (CG) and circumcoro-graphyne (CCG)) were designed with the help of First-Principles Density Functional Theory based calculations using the PBE-GGA level of theory employing a plane wave basis set. Both CG and CCG exhibit the space group p6/mmm which is akin to that of graphene. The dynamical stability of CG and CCG was analyzed by performing phonon mode analysis and molecular dynamics simulations. Interestingly, the CG shows a narrow band gap. Anisotropic Dirac cones in the proximity of the Fermi level are observed in the case of CCG. The band gap and other associated features of these novel 2D materials are sensitive to the external strain and hole/electron doping (B/N doping).
 Please wait while we load your content...
Please wait while we load your content...

