
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design of chiral urea-quaternary ammonium salt hybrid catalysts for asymmetric reactions of glycine Schiff bases†
Maximilian
Tiffner
a,
Johanna
Novacek
a,
Alfonso
Busillo
b,
Katharina
Gratzer
a,
Antonio
Massa
*b and
Mario
Waser
*a
aInstitute of Organic Chemistry, Johannes Kepler University Linz, Altenbergerstraße 69, 4040 Linz, Austria. E-mail: mario.waser@jku.at; Tel: +43 732 2468 8748
bDepartment of Chemistry and Biology, University of Salerno, 84084-Fisciano, Italy. E-mail: amassa@unisa.it; Tel: +39 089 969565
First published on 28th August 2015
Abstract
Bifunctional chiral urea-containing quaternary ammonium salts can be straightforwardly synthesised in good yield and with high structural diversity via a scalable and operationally simple highly telescoped sequence starting from trans-1,2-cyclohexanediamine. These novel hybrid catalysts were systematically investigated for their potential to control glycine Schiff bases in asymmetric addition reactions. It was found that Michael addition reactions and the herein presented aldol-initiated cascade reaction can be carried out to provide enantiomeric ratios up to 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 and good yields under mild conditions at room temperature.
:5 and good yields under mild conditions at room temperature.
Introduction
Chiral onium salt (phase-transfer) catalysis is one of the fundamental non-covalent activation strategies in asymmetric catalysis.1 Besides monofunctional chiral ammonium salt catalysts, the use of bifunctional derivatives has emerged as a powerful strategy for numerous applications.2 Whereas the vast majority of such catalysts contain an additional OH-group as the second coordination site,2–4 the design and application of ammonium salts containing alternative H-bonding motifs has so far been less exhaustively investigated.2,5–7 While the groups of Lassaletta and Fernández,5a Dixon,5b Smith,5c and Lin and Duan5d introduced powerful Cinchona alkaloid-based (thio)-urea containing bifunctional ammonium salt catalysts, Zhao et al.6 recently developed a modular approach to access α-amino acid-based (thio)-urea/ammonium salt hybrid catalysts. Simultaneously, our groups introduced trans-1,2-cyclohexanediamine-based bifunctional ammonium salts 1 (Scheme 1).7 An extensive screening of different salts allowed us to identify 1a as the most efficient chiral catalyst for asymmetric α-fluorination of β-ketoesters 2.7a In addition, catalyst 1b was found to be promising for a newly developed aldol-initiated cascade reaction of glycine Schiff base 5 with cyanobenzaldehyde 6.7b In the initial optimisation of this powerful transformation only one cyclohexanediamine-based catalyst 1 was tested and it was also found that the Cinchona alkaloid-based catalyst 9 introduced by Dixon5b and Smith5c gave the highest e.r. of 85:15 among the existing bifunctional ammonium salt motifs tested so far (Scheme 1, lower reaction).7b
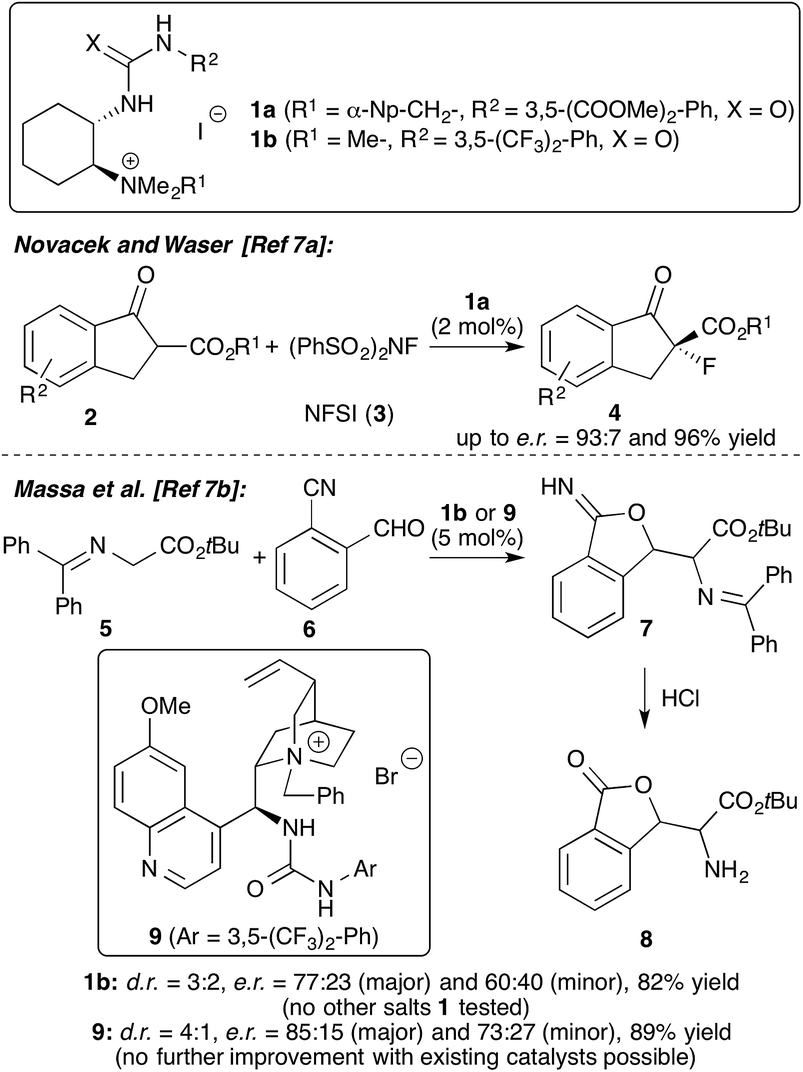 | ||
| Scheme 1 Hybrid ammonium salts 1 and their applications reported previously. | ||
These encouraging initial results with this novel family of asymmetric catalysts prompted our groups to initiate a joint project focusing on the further development and exploration of the hybrid salts 1 for stereoselective transformations. Thus we especially focused on optimisation of the existing synthesis route7a to fuel the demand for larger quantities of the readily tuneable catalysts 1 and on systematic testing of these catalysts for asymmetric reactions of glycine Schiff bases 5 (i.e. the already mentioned cascade reaction and Michael addition reactions). Hereby a detailed screening of these catalysts to increase the selectivity in the cascade synthesis of compound 8 was considered to be particularly interesting, as our modular catalyst synthesis approach should allow us to overcome the limitations of the existing catalyst motifs. Interestingly, the groups of Liu and Soloshonok recently achieved a highly asymmetric synthesis of analogous lactams in a complementary approach by employing an asymmetric auxiliary-based approach using chiral glycine Schiff base Ni(II) complexes.8 This report once again proves the high potential of this robust auxiliary approach for the synthesis of chiral α-amino acid derivatives.9
Results and discussion
Catalyst synthesis
In contrast to recent reports by others who described incorporation of the H-bonding donor first, followed by a final quaternarization step,5,6 we have chosen to use an opposite assembly strategy, carrying out the quaternarization of mono-protected diamine 11 first, followed by deprotection and coupling with an iso(thio)cyanate (Scheme 2).7a This strategy allowed us to overcome some of the challenges of the commonly used protocols,5,6 as side reactions of the nucleophilic heteroatoms of the H-bonding donor with the alkylating agent can be avoided, thus resulting in a broader and more functional group tolerant synthesis route. | ||
| Scheme 2 Catalyst synthesis: (A) R1CHO (1 eq.), NaBH4 (1.5 eq.), MeOH/THF, r.t., 15 h (no purification, >90% conversion); (B) MeI (6 eq.), K2CO3 (1.1 eq.), DMF, 60 °C, 30 h (no purification, around 70% crude yield over 2 steps); (C) CH2O (2 eq.), Na(OAc)3BH (2 eq.), DCE, r.t., 15 h (no purification, >90% conversion); (D) R1CH2Y, DMF, 60 °C, 30 h (no purification, 60–80% crude yield over 2 steps); (E) HI (10 eq.), DCM, r.t., 2 h (extractive work-up with Na2CO3); (F) R2NCX (1.2 eq.), DCM, r.t., 2–6 h (simple purification by column chromatography, 60–80% yield over 2 steps). X = O or S and Y = I, Br or Cl. | ||
As outlined in Scheme 2, the catalysts 1 can be obtained from the known Boc-protected diamine 1110 in four chemical steps. First, the introduction of bulkier (aromatic) residues for R1 on the ammonium side can be easily accomplished by means of a reductive amination, giving the corresponding secondary amines 12 in high yields (>90% conversion) and without the need for any further purification. However, the subsequent quaternarization (step B, Scheme 2) was found to be the major bottleneck in the initial protocol.7a It was not possible to introduce any residues that are sterically more demanding than a methyl group, thus allowing the syntheses of dimethyl-containing ammonium salts 13 only. In addition, we found that this exhaustive methylation is strongly dependent on the nature of substituent R1. While smaller, electron rich or electron neutral groups like a phenyl group allowed us to obtain 13 in slightly more than 50% yield starting from 11, electron-poor or sterically more demanding groups performed significantly worse (down to around 25% yield). Hereby we faced two problems: first the final quaternarization was rather slow, leading to mixtures of tert-amine intermediates and the quaternary ammonium salts even after long reaction times. In addition, under the rather forcing and prolonged conditions we observed formation of large quantities of the trimethyl ammonium salt 13b (R1 = H). Formation of the latter can be explained by a nucleophilic substitution of the benzylic ammonium groups of compound 13 (e.g. by the base or the iodide counter anion) under the harsh reaction conditions, giving the more reactive dimethyl-containing tert-amine, which was then further methylated. This effect lowered the overall yields significantly and also made purification of the ammonium salts rather difficult. In addition, when using sterically even more demanding R1 residues like an anthracenyl group no product 13 could be obtained.
To overcome these significant limitations, we first tested alternative (sterically less demanding) N-protecting groups (e.g. alloc) but with no success. Also other methylation agents did not improve the outcome. Gratifyingly, after a very careful screening of different conditions we finally found that carrying out the reaction with MeI (6 eq.) and solid K2CO3 (1.1 eq.) in DMF (60 °C) gave the target ammonium salts (13) in reliably high yields (around 70% in situ) and sufficient purity to be telescoped further in the sequence without any purification. It is noteworthy that almost no tert-amine intermediates and no trimethyl ammonium salt 13b were formed (traces of 13b originating from methylation of remaining 11 could be separated in the next step). This strategy was found to be very robust for when R1 is electron-rich, -neutral, and -poor, giving access to derivatives that were not accessible by the initial route (e.g. 3,5-(CF3)–C6H3–). Unfortunately, when using sterically demanding R1 groups (e.g. naphthyl or ortho-substituted aromatics) this route was still not very satisfactory (hereby the reactions mainly stalled at the tert-amine intermediates). However, the ammonium salts could be obtained in reasonable yields by first carrying out the dimethylation of 11 (step C) and then a final quaternarization with the appropriate benzylic halide (step D). Again the products were obtained in sufficient purity for direct further use. Thus, these newly developed quaternarization conditions allowed us to obtain a much more diverse assembly of ammonium salts 13 which were then transformed into the catalysts 1 in two more steps. The only limitations in the present synthesis are the fact that only one sterically demanding group R1 can be introduced and that groups bigger than a naphthyl group (e.g. anthracenyl) can only very slowly be incorporated.
The Boc-deprotection (step E) was initially carried out with TFA and the resulting salt (obtained by evaporation of the TFA and the solvent) was directly used for the final coupling step. However, this deprotection procedure gave mixtures of ammonium trifluoroacetates and iodides, which showed different catalytic properties and were difficult to separate. Use of HI for the deprotection was found to be beneficial for this reaction and, accompanied with an extractive work up, yielded the free amines 14 in good purity and yield. The extraction also allowed us to remove residual trimethyl ammonium salt 14b (R1 = H), which is significantly more hydrophilic than the aryl-containing derivatives. With this optimized procedure in hand, the final coupling could be carried out straightforwardly with different iso(thio)cyanates, requiring only one final (and simple) purification by silica gel column chromatography. Thus, a diversified assembly of different catalysts 1 can now be obtained in a telescoped and operationally simple manner and with satisfying overall yields (>30% based on 10) on a practical scale (up to 3 mmol).
Asymmetric reactions of the glycine Schiff bases
The first transformation that we carefully investigated was the Michael addition of glycine Schiff base 5 to acceptors like methyl acrylate 15a. This has been a thoroughly investigated reaction in asymmetric non-covalent organocatalysis in the past and it is noteworthy that most of the reported catalysts have their own characteristics with respect to their application scope (especially when using β-substituted acceptors).1,11–13 Thus we were curious to see whether our hybrid catalysts can be used (and systematically optimised) to control this important transformation. Table 1 gives an overview of the most significant results obtained from a very detailed screening of different catalysts and reactions conditions. We first identified the combination of toluene and solid Cs2CO3 (1.5 eq.) as the best-suited solvent–base system for this reaction. Larger amounts of base favoured the racemic background reaction and non-aromatic solvents or aqueous (alternative) bases generally gave significantly lower selectivities (these effects were also carefully double-checked once the most active catalyst was identified).|
|
|||||||
|---|---|---|---|---|---|---|---|
| No | R1 | R2 | X | Conc.a [M] | T [°C] | Yieldb [%] | e.r.c (R:S) |
| a Based on 5. b Isolated yield. c Determined from HPLC using a chiral stationary phase.11 d Using 5 mol% catalyst. | |||||||
| 1 | H | Ph | O | 0.17 | 25 | 60 | 67:33 |
| 2 | S | 69 | 60:40 |
||||
| 3 | Ph | O | 90 | 76:24 |
|||
| 4 | α-Np | 72 | 78:22 |
||||
| 5 | β-Np | 35 | 74:26 |
||||
| 6 | 4-tBu–C6H4 | 6 | 79:21 |
||||
| 7 | 4-NO2–C6H4 | 40 | 74:26 |
||||
| 8 | 3,5-(CF3)2–C6H3 | 85 | 84:16 |
||||
| 9 | 3-NO2–C6H4 | 83 | 81:19 |
||||
| 10 | 3,5-(CO2Me)2–C6H3 | 85 | 75:25 |
||||
| 11 | 3,5-(CF3)2–C6H3 | 95 | 75:25 |
||||
| 12 | Cy | 88 | 87:13 |
||||
| 13 | 40 | 90 | 81:19 |
||||
| 14 | 0 | 70 | 92:8 |
||||
| 15 | −20 | 5 | 92:8 |
||||
| 16 | S | 25 | 91 | 65:35 |
|||
| 17 | tBu | O | 94 | 85:15 |
|||
| 18 | Et | 96 | 89:11 |
||||
| 19 | 0.08 | 87 | 90:10 |
||||
| 20 | 0 | 45 | 95:5 |
||||
| 21 | 3,5-(CF 3 ) 2 –C 6 H 3 | Et | O | 0.02 | 25 | 85 |
95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :5 :5
|
| 22d | 74 | 88:12 |
|||||
| 23 | 0.01 | 8 | 95:5 |
||||
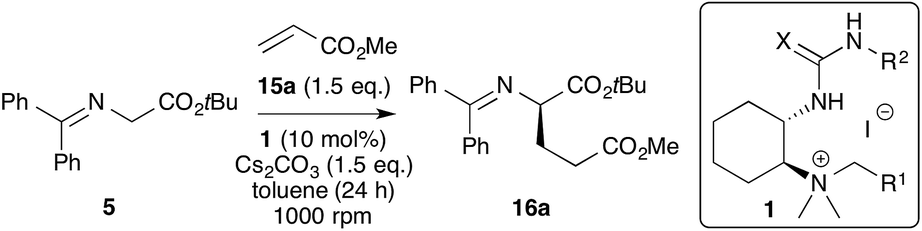
With the solvent and base conditions set, we focused on the identification of the most active catalyst. All the initial reactions were run for 24 h using the same setup (Schlenk flask, stirring rate, and dilution) and stoichiometric ratio of the reagents to assure reproducible and comparable biphasic reaction conditions.
It immediately became apparent that ureas are better-suited than thioureas (see entries 1 and 2) and that the use of benzylic ammonium salts is beneficial (entry 3 vs. 1). Thus a series of hybrid catalysts with different aryl groups (R1) on the ammonium side (keeping the phenyl-urea group R2 unchanged) were tested under identical conditions (entries 3–8 give the most significant results). Contrary to our results obtained in the α-fluorination of ketoesters7a (shown in Scheme 1), the introduction of bulky naphthyl groups as R1 was found to have no beneficial effect. In contrast, increasing the bulk by incorporating a t-butyl group on the aryl moiety (entry 6) significantly reduced the reaction rate. Also, the introduction of nitro substituents did not allow us to increase the selectivity (entry 7). Luckily, as already discussed above, the new synthesis protocol (Scheme 2) allowed us to introduce trifluoromethyl-substituted aryl groups as R1 (i.e. 3,5-(CF3)2–C6H3) in good yield. This catalyst modification turned out to be the most fruitful amongst all the tested R1 groups, giving 16a with reasonable selectivity (e.r. = 84:16) and in high yield under the standard conditions (entry 8). Based on this encouraging result, we next systematically modified the urea-substituent R2. The presence of electron-withdrawing groups led to reduced selectivities in the case of CF3-, nitro-, or diester-containing R2 groups (entries 9–11). It is noteworthy that the latter was found to be the urea-modification of choice in our recent α-fluorination protocol.7a Accordingly, these results show once more that such reactions always require a detailed and systematic screening of differently modified catalysts, thus illustrating the need for a flexible and functional group-tolerant catalyst synthesis as outlined in Scheme 2.
Interestingly, at this stage we found that replacing the aryl moieties on the urea side by incorporating a cyclohexyl group as R2 instead resulted in a good selectivity of 87:13 (entry 12). Based on the fact that aliphatic groups were never found to be promising for any of our previously investigated reactions (published7a or unpublished) this result came as a big surprise. By testing the influence of the reaction temperature we found that the selectivity can be increased by lowering the temperature, however this is accompanied by a reduced reaction rate (entries 14 and 15). Again, the reduced selectivity when using thiourea-moieties was proven by testing the analogous cyclohexyl-thiourea catalyst (entry 16). Finally, additional modifications introducing different alkyl groups as R2 allowed us to identify an ethyl group as the most powerful urea-substituent, giving 16a with an enantiomeric ratio of 89:11 under the standard conditions at room temperature. Further fine-tuning of the reaction conditions showed that the selectivity increases with higher dilution, however this results in a stepwise decrease of conversion (entries 18–23). The highest selectivities were obtained either by carrying out the reaction at a 0.02 M concentration of 5 at 25 °C (entry 21) or at higher concentration (0.08 M) at 0 °C (entry 20), albeit with reduced conversion. Furthermore, lowering the catalyst loading resulted in a reduced catalytic performance (entry 22), and further dilution did not allow us to improve the selectivity any further (entry 23).
To prove the necessity of the bifunctional nature of the catalysts 1 we also tested the simplified catalysts 17 and 18, which performed with much less selectivity or did not give any product at all under the optimized reaction conditions (Scheme 3).
 | ||
| Scheme 3 Control experiments using the simplified catalysts 17 and 18. | ||
Having identified the best-suited catalyst and the optimum reaction conditions (see entry 21, Table 1) we next investigated the application scope for this reaction (Scheme 4). One important fact that should be mentioned is that this biphasic transformation was found to be rather sensitive (e.r. and conversion) with respect to changes in the reaction setup (i.e. shape of the Schlenk flasks used and stirring rate). Therefore, when investigating the application scope we always carried out each test reaction on a 0.1 mmol scale (based on 5) with exactly the same setup and for comparison also on a smaller scale (0.02 mmol). All the results shown in Scheme 4 were thus carefully double-checked and found to be reproducible in at least two runs with two different setups each.
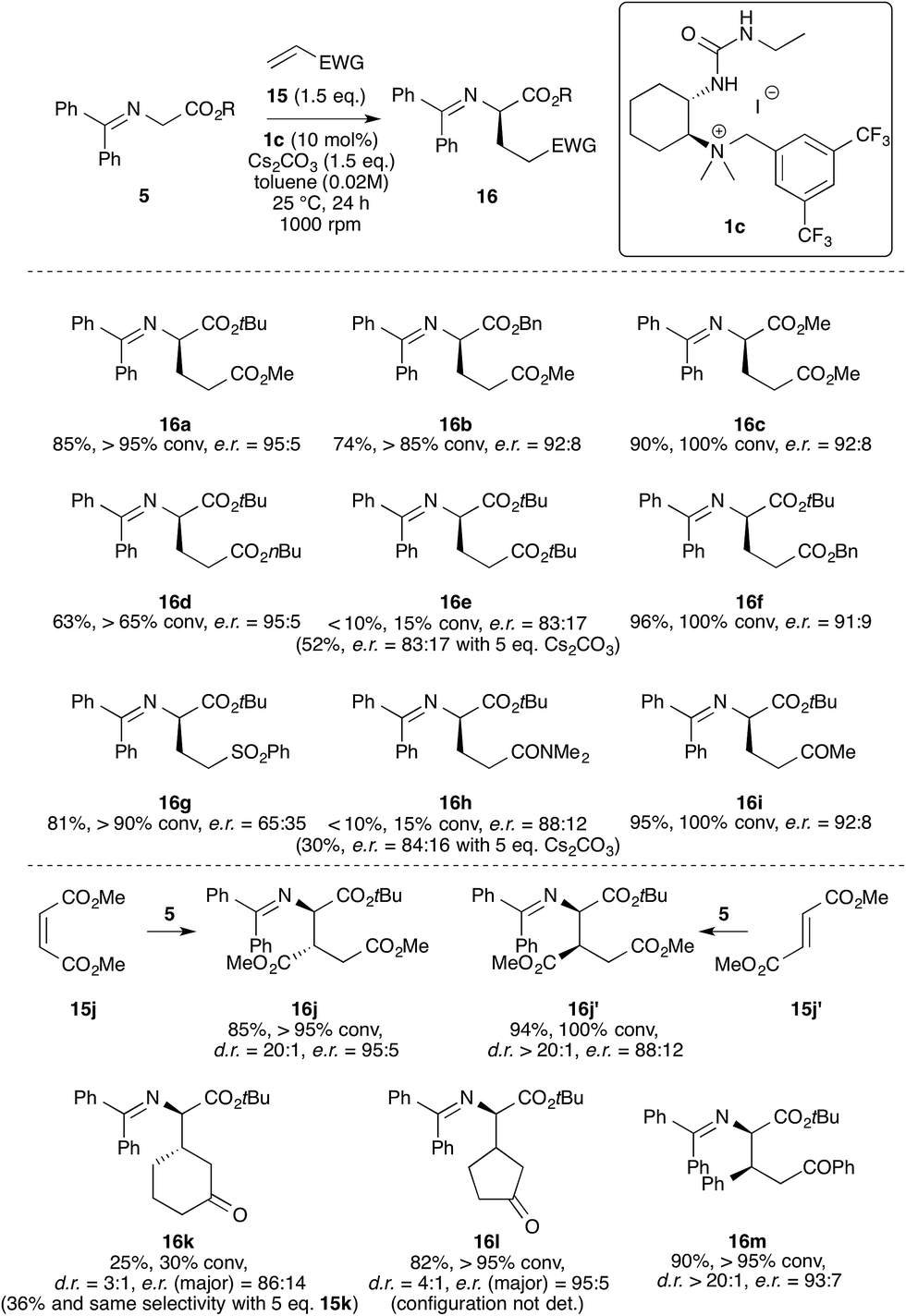 | ||
| Scheme 4 Application scope for the Michael addition of the glycine Schiff bases 5 to different Michael acceptors (15) using bifunctional catalyst 1c. The configuration of products 16a, 16b, 16d–16g, 16i, 16j′, 16k and 16m was determined by comparison of the analytical details with literature data,11–13,16,17 whereas the other products (16c, 16h, 16j and 16l) were assigned by analogy. | ||
Testing different esters of the glycine Schiff base 5 first, we found that all gave good conversion within 24 h, but that the selectivity depends on the nature of the ester group, with bulky t-butyl esters of 5 giving the highest enantioselectivity in the addition to methyl acrylate 15a (see results for products 16a–16c). Interestingly, by changing the ester group of the Michael acceptor 15 both the conversion rate14 and the enantioselectivity were influenced (see products 16d–16f). This was most notable in the synthesis of the di-t-butyl containing diester 16e (only 15% conversion under standard conditions and a reduced e.r. of 83:17). Here the conversion could be increased by using 5 equivalents of base without affecting the e.r. (when using other acceptors the use of a larger excess of base usually resulted in a reduced e.r. because of an increased rate of the racemic background reaction). When testing phenyl vinyl sulfone as an acceptor (giving product 16g) the selectivity dropped significantly, whereas N,N-dimethyl acrylamide and methyl vinyl ketone could be employed with reasonable selectivities (16h and 16i). Unfortunately, the acrylamide acceptor was found to react rather slowly and in this case the use of more base did not really allow us to overcome this limitation as the selectivity was clearly affected (this substrate was also very difficult to control in an enantioselective manner in a recent project using structurally different TADDOL-based PTCs11).
Interesting results were obtained using prochiral electrophiles 15j–15m (as shown in the lower part of Scheme 4). Using dimethyl maleate 15j, the product 16j could be obtained in good yield and with high diastereo- and enantioselectivity. It is noteworthy that Lambert and co-workers recently found that this substrate was unreactive when using their otherwise very powerful and highly selective cyclopropenimine chiral base catalysts.12 Using dimethyl fumarate 15j′ instead gave the diastereomeric 16j′ in good yield and diastereoselectivity albeit with a slightly lower enantioselectivity. Another striking difference to the chiral cyclopropenimine base catalysis was also observed when employing s-trans acceptors 15k and 15l. Although cyclohexenone 15k underwent a slow 1,4-addition under the standard conditions (which can be explained by a competing dimerization under basic phase-transfer conditions15), the stereoselectivity obtained is still reasonably high.16 Even more interestingly, when using cyclopentenone 15l we were able to obtain product 16l in high yield and with good stereoselectivity.17 Finally, chalcone was also accepted well in this reaction, providing the product 16k with reasonable selectivity and in good yield. Here it is of course fair to mention that for this substrate again Lambert’s chiral base catalyst was reported to be more selective12 and therefore the results obtained hereby may illustrate that our catalysts can provide a complementary and useful activation platform for future asymmetric organocatalytic transformations especially for addition reactions to s-trans Michael acceptors.
Having shown that the catalysts 1 can be systematically fine-tuned to obtain high selectivities for the Michael addition of the Schiff bases 5 to different Michael acceptors 15, we next addressed the recently developed aldol-initiated cascade reaction of 5 with cyanobenzaldehyde 6.7b As already discussed above, the initial screening of existing catalysts showed that the Cinchona alkaloid-based bifunctional ammonium salt 9 introduced by Dixon5b and Smith5c gave the highest selectivity with a d.r. = 4:1 and an e.r. of 85:15 for the major diastereomer (see Scheme 1). With our optimized synthesis for the catalysts 1 in hand we put our efforts into identifying an even more selective catalyst for this powerful transformation. Table 2 gives an overview of the most significant results obtained thereby. The screening was carried out under the previously developed conditions7b using solid K2CO3 as the base in CH2Cl2 (other solvents and bases were tested too but found to be less useful). In addition, initial tests showed that again ureas are more active and selective than thioureas and thus the optimization was carried out with ureas only.
|
|
||||||
|---|---|---|---|---|---|---|
| No | R1 | R2 | t [h] | Yielda [%] | d.r.b | e.r. major (minor)c |
| a Isolated yield of 8. b Determined from the 1H NMR of the crude product. c Determined from HPLC using a chiral stationary phase.7b | ||||||
| 1 | H | 3,5-(CF3)2–C6H3 | 24 | 82 | 1.5:1 |
77:23 (60:40) |
| 2 | 3-NO2–C6H4 | 24 | 66 | 1.4:1 |
77:23 (60:40) |
|
| 3 | 4-CF3–C6H4 | 48 | 63 | 1.3:1 |
76:24 (51:49) |
|
| 4 | Ph | 48 | 52 | 1.2:1 |
80:20 (59:41) |
|
| 5 | Ph | 48 | 52 | 1:1 |
77:23 (52:48) |
|
| 6 | Cy | 72 | 68 | 2:1 |
67:33 (51:49) |
|
| 7 | 4-CF3–C6H4 | 48 | 60 | 1:1 |
80:20 (57:43) |
|
| 8 | β-Np | Ph | 48 | 52 | 1:1 |
77:23 (54:46) |
| 9 | 3,5-F2–4-MeO–C6H2 | Ph | 96 | 13 | 1.1:1 |
70:30 (57:43) |
| 10 | 4-MeO–C6H3 | 3-NO2–C6H4 | 48 | 59 | 1.3:1 |
77:23 (59:41) |
| 11 | 3-NO2–C6H4 | 3-NO2–C6H4 | 26 | 65 | 2.4:1 |
85:15 (63:37) |
| 12 | 3,5-(CF 3 ) 2 –C 6 H 3 | 3-NO 2 –C 6 H 4 | 24 | 83 |
4:1
|
95:5 (75:25)
|
| 13 | 3,5-(CF3)2–C6H3 | 3,5-(CF3)2–C6H3 | 24 | 79 | 3:1 |
92:8 (73:27) |

Entry 1 shows the initially reported result7b using the trimethylammonium-based catalyst 1b and it was soon found that variation of the urea group alone does not significantly change the catalytic performance (entries 1–4). Introducing electron-neutral or bulky aryl groups on the ammonium side (entries 5–8) did not result in any improvement either. In contrast, the introduction of a naphthyl group even reduced the diastereo- and enantioselectivity (entry 8). Also the presence of aliphatic groups on the urea side (which was beneficial for our Michael reaction) did not allow us to improve the catalyst performance (entry 6) and a similar selectivity was obtained upon incorporation of more electron-rich aryl groups as R1 (entries 9 and 10).
In sharp contrast, the introduction of electron-withdrawing groups on the aryl group R1 on the ammonium side accompanied by the presence of an electron-poor aryl moiety R2 on the urea side resulted in significantly superior catalysts (entries 11–13). First, the dinitro-substituted catalyst used in entry 11 almost matched the result previously obtained with the Cinchona catalyst 9, both in terms of reactivity and selectivity. Gratifyingly, as already observed for the Michael addition, the introduction of a di-trifluoromethyl-phenyl group as R1 again significantly improved the yield and the selectivity in this transformation (entries 12 and 13). Modifying the R2 group finally resulted in the most promising catalyst, shown in entry 12, which gave product 8 in 83% isolated yield and with a good d.r. of 4:1 and excellent enantioselectivity for the major diastereoisomer (e.r. = 95:5; the minor diastereoisomer was always obtained with a lower enantiomeric ratio and no further improvement was possible).
Determination of the relative and absolute configuration of 8 turned out to be rather difficult as both diastereomers only crystallized as amorphous solids and thus X-ray structural analysis was not possible (the same was the case for the other derivatives shown in Scheme 5). The coupling constants of 7 and 8 also did not enable us to unambiguously determine the relative configuration (NOESY experiments were also not clear). However, it was possible to assign the absolute configuration of the amino acid stereogenic center of the major diastereomer to be (R) by analysis of the Mosher amide derivatives of 8a.18 Subsequent NMR studies of the compounds then indicated that the major diastereomer is most probably the anti isomer as depicted in Table 2.19
 | ||
| Scheme 5 Use of the cyanobenzaldehydes 6 for the cascade reaction. | ||
With the most active catalyst 1d in hand we quickly tested two halide- and one methoxy-containing cyanobenzaldehydes (6) under the same conditions and found that these substrates were also well tolerated, albeit the stereoselectivities slightly decreased in these cases, especially for the minor diastereoisomers (Scheme 5). Nevertheless, a first proof-of-principle for the generality of this reaction was clearly made, thus illustrating the potential of these catalysts for such cascade-reactions and providing a complementary approach to the recently reported analogous auxiliary-based protocol.8
Experimental
General information
1H and 13C-NMR spectra were recorded on a Bruker Avance III 300 MHz spectrometer, on a Bruker Avance III 700 MHz spectrometer with a TCI cryoprobe or on Bruker DRX 400, 300 or 250 MHz spectrometers. All NMR spectra were referenced to the solvent peak. High resolution mass spectra were obtained using an Agilent 6520 Q-TOF mass spectrometer with an ESI source and an Agilent G1607A coaxial sprayer or using a Thermo Fisher Scientific LTQ Orbitrap XL with an Ion Max API Source. Additional mass spectral analyses were carried out using an electrospray spectrometer, a Waters 4 micro quadrupole. Elemental analyses were performed with FLASHEA 1112 series-Thermo Scientific for CHNS-O apparatus. IR spectra were recorded on a Bruker Tensor 27 FT-IR spectrometer with an ATR unit. HPLC analyses were performed either by using a Waters instrument or using a Dionex Summit HPLC system with a Chiralcel OD-H (250 × 4.6 mm, 5 μm), a Chiralcel OD-R (250 × 4.6 mm, 10 μm), or a Chiralpak AD-H (250 × 4.6 mm, 5 μm) chiral stationary phase. Optical rotations were recorded on a PerkinElmer Polarimeter Model 241 MC and on a Schmidt + Haensch Polarimeter Model UniPol L 1000. All chemicals were purchased from commercial suppliers and used without further purification unless otherwise stated. All reactions were performed under an Ar atmosphere.A detailed experimental section including all the procedures and analytical data of the catalysts and asymmetric reaction products as well as copies of the NMR spectra and HPLC chromatograms can be found in the ESI.†
Synthesis of catalysts 1c and 1d
:1) and the solution was stirred at r.t. overnight. After addition of NaBH4 (114 mg, 3 mmol, 1.5 eq.), stirring was continued for another 2 h at r.t. The reaction was quenched by addition of water and extracted with water/diethyl ether. The organic phase was washed with brine, dried over Na2SO4, and evaporated to dryness to obtain the crude product 12c in almost quantitative yield (95%, 824 mg, 1.9 mmol), which could be directly used without any purification. Compound 12c (R1 = 3,5-(CF3)2–C6H3): 1H NMR (300 MHz, δ, CDCl3, 298 K): 1.08–1.35 (m, 4H), 1.42 (s, 9H), 1.61–1.85 (m, 2H), 1.96 (s (b), 1H), 1.99–2.15 (m, 2H), 3.31–3.47 (m, 1H), 3.83 (d, 1H, J = 14.1 Hz), 4.00 (d, 1H, J = 14.1 Hz), 4.48 (d, 1H, J = 7.3 Hz), 7.73 (s, 1H), 7.82 (s, 2H) ppm.
![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) = 3431, 3270, 3011, 2980, 2939, 2867, 1695, 1625, 1516, 1467, 1455, 1393, 1370, 1323, 1281, 1242, 1176, 1138, 1048, 1024, 904, 870, 844, 737, 709, 683 cm−1; HRMS (ESI) m/z calcd for C22H31F6N2O2+: 469.2284 [M+], found: 469.2281.
:methanol = 50:1 to 10:1) to obtain pure catalyst 1 in the reported yields.
= 3295, 3021, 2988, 2936, 2864, 2349, 2288, 1656, 1546, 1449, 1373, 1278, 1174, 1130, 904, 843, 751, 719, 682, 663, 593, 463, cm−1; HRMS (ESI): m/z calcd for C20H28F6N3O+: 440.2131 [M+]; found: 440.2118.
= 3462, 3254, 3031, 2944, 2866, 1692, 1600, 1548, 1529, 1485, 1451, 1434, 1372, 1352, 1325, 1280, 1206, 1178, 1137, 904, 843, 830, 798, 737, 709, 683 cm−1; HRMS (ESI): m/z calcd for C24H27F6N4O3+: 533.1982 [M+]; found: 533.1998.
= 3431, 3270, 3011, 2980, 2939, 2867, 1695, 1625, 1516, 1467, 1455, 1393, 1370, 1323, 1281, 1242, 1176, 1138, 1048, 1024, 904, 870, 844, 737, 709, 683 cm−1; HRMS (ESI) m/z calcd for C22H31F6N2O2+: 469.2284 [M+], found: 469.2281.
:methanol = 50:1 to 10:1) to obtain pure catalyst 1 in the reported yields.
= 3295, 3021, 2988, 2936, 2864, 2349, 2288, 1656, 1546, 1449, 1373, 1278, 1174, 1130, 904, 843, 751, 719, 682, 663, 593, 463, cm−1; HRMS (ESI): m/z calcd for C20H28F6N3O+: 440.2131 [M+]; found: 440.2118.
= 3462, 3254, 3031, 2944, 2866, 1692, 1600, 1548, 1529, 1485, 1451, 1434, 1372, 1352, 1325, 1280, 1206, 1178, 1137, 904, 843, 830, 798, 737, 709, 683 cm−1; HRMS (ESI): m/z calcd for C24H27F6N4O3+: 533.1982 [M+]; found: 533.1998.
General procedure for the asymmetric Michael reactions of 5
Degased toluene (5 mL) was added to a mixture of the Schiff base 5 (0.1 mmol), catalyst 1c (10 mol%), and Cs2CO3 (1.5 eq.) in a Schlenk tube. The stirring rate was set to 1000 rpm and the corresponding electrophile 15 (1.5 eq.) was added. After 24 h at 25 °C the reaction mixture was filtered through a plug of Na2SO4. The solvent was removed under reduced pressure. The crude products were purified by column chromatography (silica gel, heptanes:EtOAc = 20:1 to 2:1) giving the Michael addition products (16) in the reported yields.
:5 upon reacting Schiff base 5a with acrylate 15a in the presence of 10 mol% 1c at 25 °C under the general procedure conditions. The analytical data are in full accordance with those reported in the literature.11,12 [α]21D (c = 0.70, CHCl3) = 74.9°; 1H NMR (300 MHz, δ, CDCl3, 298 K): 1.44 (s, 9H), 2.16–2.27 (m, 2H), 2.33–2.41 (m, 2H), 3.59 (s, 3H), 3.93–3.99 (m, 1H), 7.14–7.21 (m, 2H), 7.28–7.47 (m, 6H), 7.60–7.68 (m, 2H) ppm; 13C NMR (75 MHz, δ, CDCl3, 298 K): 28.2, 28.8, 30.5, 51.7, 64.9, 81.3, 127.9, 128.1, 128.6, 128.7, 128.9, 130.5, 136.6, 139.6, 170.8, 170.9, 173.7 ppm; IR (film): = 2978, 2926, 1738, 1707, 1661, 1599, 1578, 1449, 1369, 1319, 1279, 1260, 1234, 1153, 943, 920, 849, 812 cm−1; the enantioselectivity was determined using HPLC (Chiralcel AD-H, eluent: n-hexane:i-PrOH = 95:5, 0.5 mL min−1, 10 °C, retention times: 10.9 min (major; R-enantiomer), 12.3 min (minor; S-enantiomer)); the absolute configuration was determined by comparison of the retention times and [α]D value with literature data.12 HRMS (ESI): m/z calcd for C23H27NO4: 382.2013 [M + H]+; found: 382.2013.
General procedure for the cascade reactions of 5
In a round-bottom flask, a 2-cyanobenzaldehyde 6 (0.10 mmol) was added at room temperature to a stirred solution of glycine Schiff base 5 (1.1 eq., 0.11 mmol), K2CO3 (1 eq.), and catalyst 1d (5% mol) in CH2Cl2 (3 mL). The mixture was stirred at r.t. for 24 h (1000 rpm). Afterwards, the mixture was purified directly using flash chromatography on silica gel with hexane:ethyl acetate = 8:2 to give the intermediate 7 as a mixture of diastereoisomers. The products (7) were dissolved in a cooled solution of 0.5 M HCl (1 mL) and THF (3 mL) (0 °C). The mixture was stirred at the same temperature for 2 h and then concentrated under vacuum. The resulting residue was treated with saturated NaHCO3 (20 mL), extracted with CH2Cl2 (4 × 30 mL), and purified using flash chromatography (silica gel, hexanes:EtOAc = 2:1).
:1 and e.r. = 95:5 for the major diastereoisomer. [α]20D (c = 0.5, CHCl3) = −2.5°. 1H-NMR (300 MHz, δ, CDCl3, 298 K): 1.40 (s, 9H), 1.61 (br s, 2H), 4.04 (d, J = 3.6 Hz, 1H), 5.77 (d, J = 3.5 Hz, 1H), 7.44 (d, J = 7.6 Hz, 1H), 7.58 (t, J = 7.42 Hz, 1H), 7.68 (t, J = 6.48 Hz, 1H), 7.92 (d, J = 7.6 Hz, 1H); 13C-NMR (100 MHz, δ, CDCl3): 29.1, 58.7, 83.3, 83.9, 123.8, 126.9, 128.3, 130.8, 135.2, 147.5, 171.3, 171.5; MS (ESI): m/z = 264.1 (M + H)+; anal. calcd for C14H17NO4: C, 63.87; H, 6.51; N, 5.32. Found: C, 63.97; H, 6.41; N, 5.37%. The enantioselectivity was determined using HPLC (Chiralcel OD-H, eluent: n-hexane:i-PrOH = 90:10, 0.7 mL min−1, 25 °C, retention times (major diastereomer): 25.3 min (minor), 34.3 min (major)).
Conclusions
A flexible and highly telescoped synthesis strategy for a structurally diverse library of chiral cyclohexanediamine-based urea-containing quaternary ammonium salts was developed. The catalysts were successfully employed in asymmetric Michael addition reactions and a new powerful aldol-initiated cascade reaction of glycine Schiff bases with cyanobenzaldehyde derivatives. In both cases the flexible catalyst strategy allowed us to systematically fine-tune the catalysts for these reactions, thus resulting in high enantioselectivities and good to excellent yields. Besides the high enantioselectivities, it was also shown that these catalysts are very promising for the control of s-trans Michael acceptors, thus providing a powerful catalyst platform for further challenging asymmetric transformations.Acknowledgements
This work was supported by the Austrian Science Funds (FWF): Project No. P26387-N28. Financial support from the Federal State Government of Upper Austria (Research Fellowship to J. N.) is kindly acknowledged. We are grateful to Dr Markus Himmelsbach for his support with HRMS analysis. The NMR spectrometers used at JKU Linz were acquired in collaboration with the University of South Bohemia (CZ) with financial support from the European Union through the EFRE INTERREG IV ETC-AT-CZ program (project M00146, “RERI-uasb”).Notes and references
- For selected reviews about asymmetric phase-transfer catalysis see: (a) K. Maruoka, Asymmetric Phase Transfer Catalysis, WILEY-VCH, Weinheim, 2008 Search PubMed; (b) M. J. O’Donnell, in Catalytic Asymmetric Syntheses, ed. I. Ojima, WILEY-VCH, New York, 2nd edn, 2000, pp. 727–755 Search PubMed; (c) K. Maruoka and T. Ooi, Chem. Rev., 2003, 103, 3013–3028 CrossRef CAS PubMed; (d) M. J. O’Donnell, Acc. Chem. Res., 2004, 37, 506–517 CrossRef PubMed; (e) T. Ooi and K. Maruoka, Angew. Chem., Int. Ed., 2007, 46, 4222–4266 CrossRef CAS PubMed; (f) S. Shirakawa and K. Maruoka, Angew. Chem., Int. Ed., 2013, 52, 4312–4348 CrossRef CAS PubMed; (g) S.-S. Jew and H.-G. Park, Chem. Commun., 2009, 7090–7103 RSC; (h) R. Herchl and M. Waser, Tetrahedron, 2014, 70, 1935–1960 CrossRef CAS PubMed.
- For a review about bifunctional chiral ammonium salt-based catalysts see: J. Novacek and M. Waser, Eur. J. Org. Chem., 2013, 637–648 CrossRef CAS PubMed.
- For pioneering reports using Cinchona alkaloid-based free OH-containing ammonium salt catalysts see: (a) R. Helder, J. C. Hummelen, R. W. P. M. Laane, J. S. Wiering and H. Wynberg, Tetrahedron Lett., 1976, 17, 1831–1834 CrossRef; (b) U. H. Dolling, P. Davis and E. J. J. Grabowski, J. Am. Chem. Soc., 1984, 106, 446–447 CrossRef CAS; (c) M. J. O’Donnell, W. D. Bennett and S. Wu, J. Am. Chem. Soc., 1989, 111, 2353–2355 CrossRef.
- For selected very recent examples describing the beneficial use of free OH-containing chiral ammonium salt catalysts see: (a) J. R. Wolstenhulme, A. Cavell, M. Gredicak, R. W. Driver and M. D. Smith, Chem. Commun., 2014, 50, 13585–13588 RSC; (b) B. Xiang, K. M. Belyk, R. A. Reamer and N. Yasuda, Angew. Chem., Int. Ed., 2014, 53, 8515–8518 CrossRef PubMed; (c) R. J. Armstrong and M. D. Smith, Angew. Chem., Int. Ed., 2014, 53, 12822–12826 CrossRef CAS PubMed; (d) S. Shirakawa, L. Wang, R. He, S. Arimitsu and K. Maruoka, Chem.–Asian J., 2014, 9, 1586–1593 CrossRef CAS PubMed; (e) S. Shirakawa, T. Tokuda, S. B. J. Kan and K. Maruoka, Org. Chem. Front., 2015, 2, 336–339 RSC.
- For (thio)-urea containing Cinchona alkaloid-based ammonium salt catalysts see: (a) P. Bernal, R. Fernández and J. M. Lassaletta, Chem.–Eur. J., 2010, 16, 7714–7718 CrossRef CAS PubMed; (b) K. M. Johnson, M. S. Rattley, F. Sladojevich, D. M. Barber, M. G. Nunez, A. M. Goldys and D. J. Dixon, Org. Lett., 2012, 14, 2492–2495 CrossRef CAS PubMed; (c) M. Li, P. A. Woods and M. D. Smith, Chem. Sci., 2013, 4, 2907–2911 RSC; (d) B. Wang, Y. Liu, C. Sun, Z. Wei, J. Cao, D. Liang, Y. Lin and H. Duan, Org. Lett., 2014, 16, 6432–6435 CrossRef CAS PubMed; (e) P. G. K. Clark, L. C. C. Vieira, C. Tallant, O. Fedorov, D. C. Singleton, C. M. Rogers, O. P. Monteiro, J. M. Bennett, R. Baronio, S. Müller, D. L. Daniels, J. Mendez, S. Knapp, P. E. Brennan and D. J. Dixon, Angew. Chem., Int. Ed., 2015, 54, 6217–6221 CrossRef CAS PubMed.
- For amino acid-based (thio)-urea containing ammonium salts see: (a) H.-Y. Wang, Z. Chai and G. Zhao, Tetrahedron, 2013, 69, 5104–5111 CrossRef CAS PubMed; (b) H.-Y. Wang, J.-X. Zhang, D.-D. Cao and G. Zhao, ACS Catal., 2013, 3, 2218–2221 CrossRef CAS.
- For trans-1,2-cyclohexanediamine-based (thio)-urea containing ammonium salts developed by our groups see: (a) J. Novacek and M. Waser, Eur. J. Org. Chem., 2014, 802–809 CrossRef CAS PubMed; (b) M. Perillo, A. Di Mola, R. Filosa, L. Palombi and A. Massa, RSC Adv., 2014, 4, 4239–4246 RSC.
- T. Li, S. Zhou, J. Wang, J. L. Acena, V. A. Soloshonok and H. Liu, Chem. Commun., 2015, 51, 1624–1626 RSC.
- (a) A. E. Sorochinsky, J. L. Acena, H. Moriwaki, T. Sato and V. A. Soloshonok, Amino Acids, 2013, 45, 691–718 CrossRef CAS PubMed; (b) A. E. Sorochinsky, J. L. Acena, H. Moriwaki, T. Sato and V. A. Soloshonok, Amino Acids, 2013, 45, 1017–1033 CrossRef CAS PubMed.
- D. W. Lee, H.-J. Ha and W. K. Lee, Synth. Commun., 2007, 37, 737–742 CrossRef CAS PubMed.
- Use of TADDOL-based PTCs: G. N. Gururaja, T. Herchl, A. Pichler, K. Gratzer and M. Waser, Molecules, 2013, 18, 4357–4372 CrossRef CAS PubMed.
- Use of cyclopropenimine-based chiral base catalysts: (a) J. Bandar and T. H. Lambert, J. Am. Chem. Soc., 2012, 134, 5552–5555 CrossRef CAS PubMed; (b) J. Bandar, G. S. Sauer, W. D. Wulff, T. H. Lambert and M. J. Vetticatt, J. Am. Chem. Soc., 2014, 136, 10700–10707 CrossRef CAS PubMed; (c) J. Bandar, A. Barthelme, A. Y. Mazori and T. H. Lambert, Chem. Sci., 2015, 6, 1537–1547 RSC.
- Use of oxo-pyrimidinium-based catalysts: A. E. Sheshenev, E. V. Boltukhina, A. J. P. White and K. K. Hii, Angew. Chem., Int. Ed., 2013, 52, 6988–6991 CrossRef CAS PubMed.
- In those cases where the conversion was at least 60–70% after 24 h, stirring for another day was usually sufficient to ensure >95% conversion of starting material 5.
- R. Ceccarelli, S. Insogna and M. Bella, Org. Biomol. Chem., 2006, 4, 4281–4284 CAS.
- For other reports on the synthesis of this compound see: (a) E. J. Corey, M. C. Noe and F. Xu, Tetrahedron Lett., 1998, 39, 5347–5350 CrossRef CAS; (b) R. Chinchilla, P. Mazón, C. Nájera, F. J. Ortega and M. Yus, ARKIVOC, 2005, 222–232 CAS; (c) V. I. Maleev, M. North, V. A. Larionov, I. V. Fedyanin, T. F. Savel’yeva, M. A. Moscalenko, A. F. Smolyakov and Y. N. Belokon, Adv. Synth. Catal., 2014, 356, 1803–1810 CrossRef PubMed.
- For a highly enantioselective but only moderately diastereoselective copper-catalyzed protocol for this reaction see: M. Strohmeier, K. Leach and M. A. Zajak, Angew. Chem., 2011, 123, 12543–12546 CrossRef PubMed.
- For a comprehensive overview see: J. M. Seco, E. Quinoa and R. Riguera, Chem. Rev., 2004, 104, 17–117 CrossRef CAS.
- Detailed VCD studies to unambiguously prove this assignment will soon be carried out in collaboration with specialists.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and copies of NMR spectra and HPLC analyses. See DOI: 10.1039/c5ra14466c |
| This journal is © The Royal Society of Chemistry 2015 |