
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Room temperature synthesis of an amorphous MoS2 based composite stabilized by N-donor ligands and its light-driven photocatalytic hydrogen production†
Felix
Niefind
a,
John
Djamil
a,
Wolfgang
Bensch
*a,
Bikshandarkoil R.
Srinivasan
b,
Ilya
Sinev
c,
Wolfgang
Grünert
c,
Mao
Deng
d,
Lorenz
Kienle
d,
Andriy
Lotnyk
e,
Maria B.
Mesch
f,
Jürgen
Senker
f,
Laura
Dura
g and
Torsten
Beweries
g
aChristian-Albrechts-Universität zu Kiel, Institute of Inorganic Chemistry, 24118 Kiel, Germany. E-mail: wbensch@ac.uni-kiel.de
bGoa University, Department of Chemistry, Goa 403206, India
cRuhr-Universität Bochum, Laboratory of Industrial Chemistry, 44801 Bochum, Germany
dChristian-Albrechts-Universität zu Kiel, Institute for Materials Science, 24143 Kiel, Germany
eLeibniz Institute of Surface Modification IOM, 04318 Leipzig, Germany
fInorganic Chemistry III, University of Bayreuth, 95440 Bayreuth, Germany
gUniversität Rostock, Leibniz-Institut für Katalyse e.V. (LIKAT), 18059 Rostock, Germany
First published on 30th July 2015
Abstract
Herein an entirely new and simple room temperature synthesis of an amorphous molybdenum sulfide stabilized by complexing ammonia and hydrazine is reported. The resulting material exhibits an outstanding activity for the photocatalytic hydrogen evolution driven by visible light. It is chemically stable during the reaction conditions of the photocatalysis and shows unusual thermal stability up to 350 °C without crystallization. The new material is obtained by a reaction of solid ammonium tetrathiomolybdate and gaseous hydrazine. In the as-prepared state Mo atoms are surrounded by μ2-briding S2−, NH3 and hydrazine, the latter being coordinated to Mo(IV) in a bridging or side-on mode. Heating at 450 °C or irradiation with an electron beam generates nanosized crystalline MoS2 slabs. The two modes for crystallization are characterized by distinct mechanisms for crystal growth. The stacking of the slabs is low and the material exhibits a pronounced turbostratic disorder. Heat treatment at 900 °C yields more ordered MoS2 but structural disorder is still present. The visible-light driven hydrogen evolution experiments evidence an outstanding performance of the as-prepared sample. The materials were thoroughly characterized by optical spectroscopy, chemical analysis, in situ HRTEM, XRD, 1H and 15N solid-state NMR, XPS, and thermal analysis.
1. Introduction
Since the world's population keeps on growing, forecasted to reach the 9.5 billion mark around the middle of the century, the most pressing challenge of our time is the sufficient supply of clean energy. The use of fossil fuels is reaching its limit due to the harmful effect of combustion products upon the environment, which is shifting the focus of research towards renewable energy sources like solar or wind energy.1,2 Hydrogen generated by sunlight is widely discussed as a clean energy source which generates only water as exhaust. Since solar light is accessible in most regions on the planet the photocatalytic hydrogen production3–5 or the photoelectrochemical hydrogen evolution6–10 reaction are considered to be promising approaches. The best electrocatalysts for the hydrogen evolution reaction (HER) are platinum group metals which unfortunately are rare and expensive. Hence, these metals are not suitable for large scale applications and therefore intense research is going on worldwide to identify materials, which can replace these precious metals.Molybdenum sulfide (MoS2) has been intensely studied as possible catalysts for HER since the 1970s, most notably as nanocrystalline MoS2.11–14 Besides application of nanocrystalline MoS2 as electrocatalyst or for photoelectrochemical application in HER,15–17 particular interest has been devoted to the visible-light driven hydrogen production.18–21 In the last few years amorphous MoS2+x was identified as efficient photocatalyst for HER,22,23 which are highly active catalyst materials, often even more active than their crystalline counterpart,24–26 and they can be synthesized using electrodeposition or wet chemical reactions with no need for a thermal sulfidization treatment.27–30 Amorphous MoS2+x materials exhibit Mo(IV) centers with a local environment of S2− and S22− anions.31–34 Upon applying a cathodic potential to amorphous MoS2+x materials in the HER the surface composition changes to MoS2, as was revealed by chemical and physical characterizations.35–37 The results of these studies suggest that amorphous MoS2 is at least an important component of the catalytically active material. In the past it was shown that solution based syntheses applying (NH4)2MoS4 (ATM) as source and using e.g. hydrazine as reducing agent yield amorphous sulfur rich products MoS2+x and only if the reaction slurry was refluxed at T ≈ 90 °C poorly crystalline MoS2 with some S excess was obtained.38–45 The large excess of hydrazine always generates S22− species which are bound to the Mo center yielding MoS2+x.
These observations led to the main idea of the present work: is it possible to develop a new kinetically controlled synthetic route for the direct preparation of amorphous MoS2 avoiding formation of MoS3 or MoS2+x as intermediates, which must then be transformed to MoS2 by applying elevated temperature yielding inevitable nanocrystalline MoS2?
2. Results and discussion
2.1. Analysis of as prepared PX
Amorphous PX was synthesized by decomposition of ammonium tetrathiomolybdate (ATM) in a desiccator under hydrazine atmosphere (see Experimental section). The gaseous N2H4 molecules are proposed to be decomposed on the surface of ATM crystallites according to the reaction N2H4 → N2 + 4H+ + 4e−. The protons attack the sulfide anions forming H2S of the [MoS4]2− ion and the electrons simultaneously reduce Mo(VI) to Mo(IV). The chemical analyses of numerous syntheses yield as average composition MoS2.0(2)No.8(2)H3.3(2) for the reaction products. Obviously the amorphous products contain NH3 and may be also some residual hydrazine (see below). The presence of NH3 was verified by reacting PX with Nessler's reagent in water.X-Ray powder patterns of as prepared PX, of samples heated to 450 °C and 900 °C (PX450 and PX900) and a simulated pattern of bulk MoS2 are displayed in Fig. 1. The pattern of PX is dominated by a very broad hump between 4° and 15° 2θ and a less intense modulation at around 32–33° 2θ reminiscent of a glass-like state. The evolution of the background at low scattering angles gives also hints that no single or double-layered material was formed.46
 | ||
| Fig. 1 XRD pattern of PX (orange trace), PX after H2-production (green trace), of PX heated to 450 and 900 °C (blue and red trace), and for comparison a calculated pattern of MoS2 (black trace). | ||
Transmission electron microscopy (TEM) analysis of PX revealed several remarkable results. Specifically crystallization phenomena occurred during analysis, which are documented in Fig. 2.
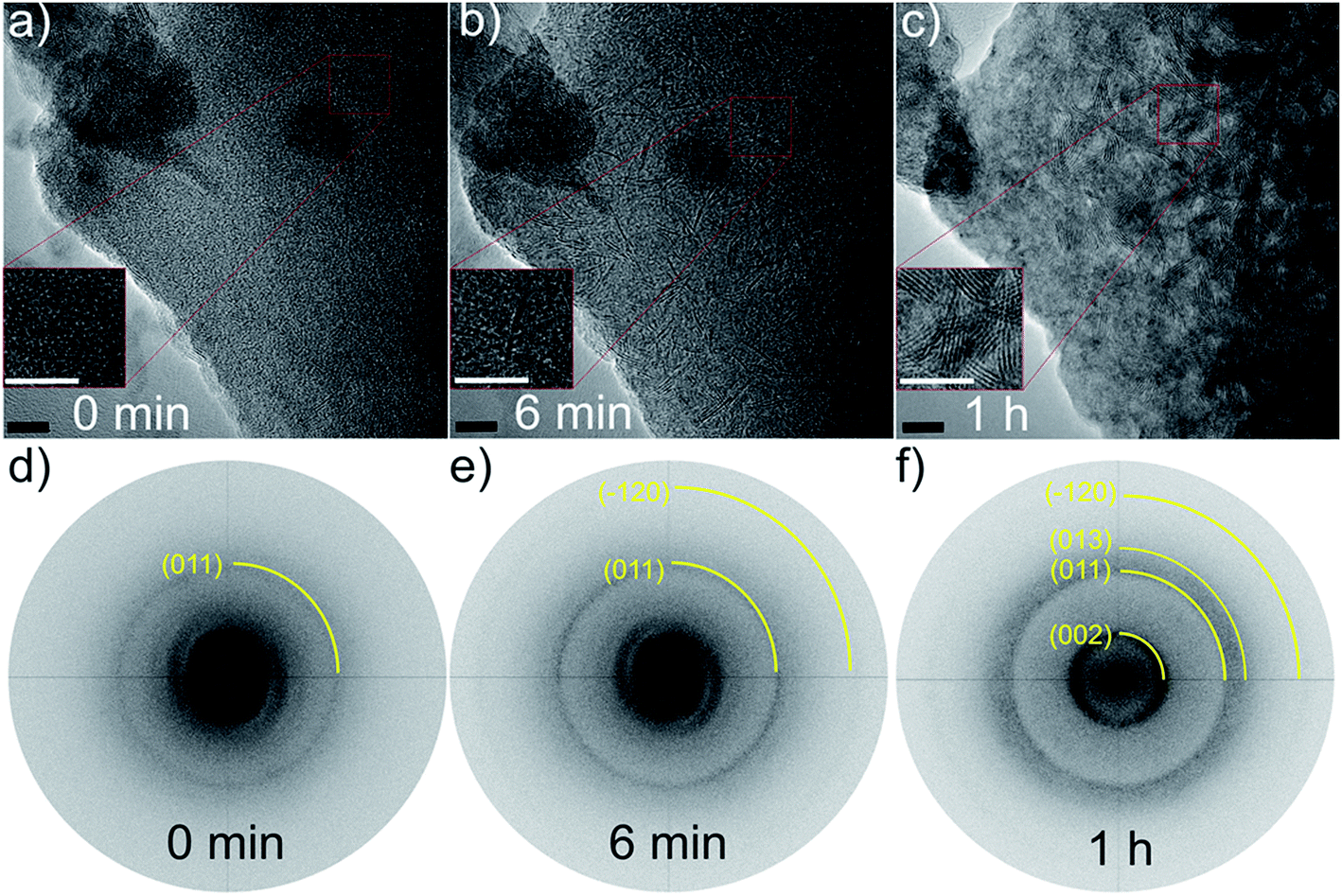 | ||
| Fig. 2 Time resolved series of HRTEM micrographs of a region of the PX showing the evolution of (002) slabs under irradiation with an electron beam voltage of 300 kV for (a) 0 min, (b) 6 min, (c) 1 h, and the corresponding Fast Fourier Transformation (FFT) patterns (d)–(f). Insets show enlarged views of the development of MoS2 slabs in the same position of the bulk material. Scale bar is 10 nm. The measured d-values of the (002): 0.637 nm, (011); 0.264 nm, (013): 0.222 nm, (−120): 0.156 nm, as compared to the theoretical values: 0.615 nm, 0.267 nm, 0.228 nm, 0.158 nm, the (002) d-spacing is larger than the theoretical one indicating a more bended and disordered arrangement of the MoS2 basal planes produced via in situ electron irradiation. | ||
For the pristine state, a combined approach of high resolution (HR)TEM/FFT (Fast Fourier Transformation) confirmed a completely amorphous structure of the sample, as depicted by the HRTEM micrograph and the corresponding FFT in Fig. 2a and (d).
For further characterization Raman spectroscopy was applied to probe the layer character of PX. The Raman active modes E12g and A1g of MoS2 representing the in-plane and out-of-plane vibrations, are typically located at about 386 and 411 cm−1, respectively (Fig. S1†).47,48 These signals differ in intensity and energy separation depending on the number of stacked MoS2 layers. The more layers are stacked the higher is the intensity of the peaks and the smaller is the energy difference between these signals. No resonances occur in the Raman spectrum of PX supporting the findings of the TEM and XRD measurements (see below).
Comparison of the FIR spectra of PX as obtained, PX450, PX900 and commercial MoS2 demonstrates that the amorphous material PX does not have a MoS2 like structure because the typical vibrations of crystalline MoS2 are absent (see Fig. S2†). The spectrum shows a band at 478 cm−1, which can be assigned to a Mo–N vibration and an absorption at 334 cm−1 which fits to the deformation vibration of a Mo–S–Mo group (see Fig. S3†). This μ2-S2− bridging ligand between two Mo atoms can be observed by FIR in MoS2 because of imperfections in the arrangement of the MoS2 sheets having defects in their basal sulfur atom arrangement.49
To get more information about the nitrogen species in the sample 15N CP MAS and 1H spin echo NMR spectra of PX as prepared were recorded (Fig. 3). The 15N NMR spectrum shows a single broad peak centered at −380 ppm with a FWHM of roughly 50 ppm, which is a result of the weak proton decoupling (see Experimental details) and distributions of the chemical shift. The observed chemical shift region between −300 and −450 ppm is consistent with expected shifts of hydrazine (−320 to −390 ppm), ammonia (around −382 ppm) and ammonium (−340 to −360 ppm).50 For hydrazine three different coordination types have to be distinguished. While for the bridging and side-on coordination in several metal–organic compounds only one signal is observed between −370 and −390 ppm, the end-on coordination reveals two peaks at around −370 ppm for the metal coordinated NH2 unit and around −320 ppm for the non-coordinated NH2 group.51 Although all three target molecules are in agreement with the 15N MAS spectral data the end-on coordination of hydrazine is unlikely since the spectral intensity around −320 ppm is low compared to the other regions.
 | ||
| Fig. 3 15N CP MAS (left) and 1H spin echo (right) NMR spectrum of PX collected at a spinning speed of 10 and 60 kHz, respectively. | ||
Further insight is provided by the high-resolution 1H NMR spectrum (Fig. 3, right). Here, one resonance around 4.34 ppm with a broad and intense shoulder at about 2.8 ppm is observed. While the former signal favors NH3 (4–7 ppm),52 or N2H4 in a side-on coordination (5–6 ppm) the latter one might be assigned to bridging (2–5 ppm) hydrazine molecules.50,53–55 Since the resonance for the non-coordinated unit of N2H4 in an end-on coordination is usually high-field shifted by about one ppm only a small amount of end-on bonded hydrazine is expected. This is in accordance with the 15N NMR results. Additionally, the low intensity in the downfield region above 7 ppm indicates a rather low concentration of ammonium (7–9 ppm).56 Combined with the elemental analysis, the NMR data favor a scenario where both NH3 and N2H4 are present in PX, with NH3 being the majority component. For N2H4 either a bridging or a side-on coordination is possible.
Since from NMR spectroscopy only the nitrogen-containing molecules could be identified XPS measurements were performed to gain more information of the chemical states of the constituents. In the survey spectra (see Fig. S5†) peaks of Mo, S, and N are clearly visible, but also those of C 1s and a very small O 1s signal caused by a slight surface contamination. Note that no Mo, S or N oxidic species can be detected in the Mo 3d, S 2p or N 1s regions (see discussion below). The surface composition of the sample derived from the XPS spectrum using the Mo 3d, S 2p and N 1s core level lines and applying the Scofield cross-section values is MoS2.1N0.8 which is very close to the data obtained by chemical analysis. For an overview of the XPS data the binding energies of the Mo 3d and S 2p fits are provided in Table 1.
Generally, the electron density of an atom, which is often related to the oxidation state, depends on several factors like number and type of ligands (covalency), difference of electronegativity as well as on the interatomic bond lengths. Thus, the Eb values reported for a distinct element in literature often scatter in a wide range depending on the actual chemical composition. Moreover, the measured Eb is affected by intra- and extraatomic relaxation and the Madelung contribution. Since PX is amorphous and contains N and H besides S, a comparison with data of crystalline bulk materials for the assignment of the species should be done with some caution.
In Fig. 4c the XPS spectrum of the N 1s and Mo 3p3/2 region is depicted. It is dominated by the Mo 3p3/2 peak at 394.5 eV, which extends into the region of the N 1s peak at 399.3 eV. In accordance with the analysis of the Mo 3d signal (see below), the Mo 3p3/2 peak was fitted with three overlapping peaks at 394.3 eV, 394.8 eV and 395.7 eV, which reproduced the signal shape perfectly. Depending on the N containing chemical compound, N 1s binding energies are located between about 407 eV (nitrate) and ca. 397 eV (nitride).57,58 Two N 1s lines at 399.3 eV and 401.3 eV (ratio: ≈8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) can be identified which can be assigned to NH3 and N2H4 coordinating the Mo center in the bridging or side-on mode, in accordance with the NMR data.
:1) can be identified which can be assigned to NH3 and N2H4 coordinating the Mo center in the bridging or side-on mode, in accordance with the NMR data.
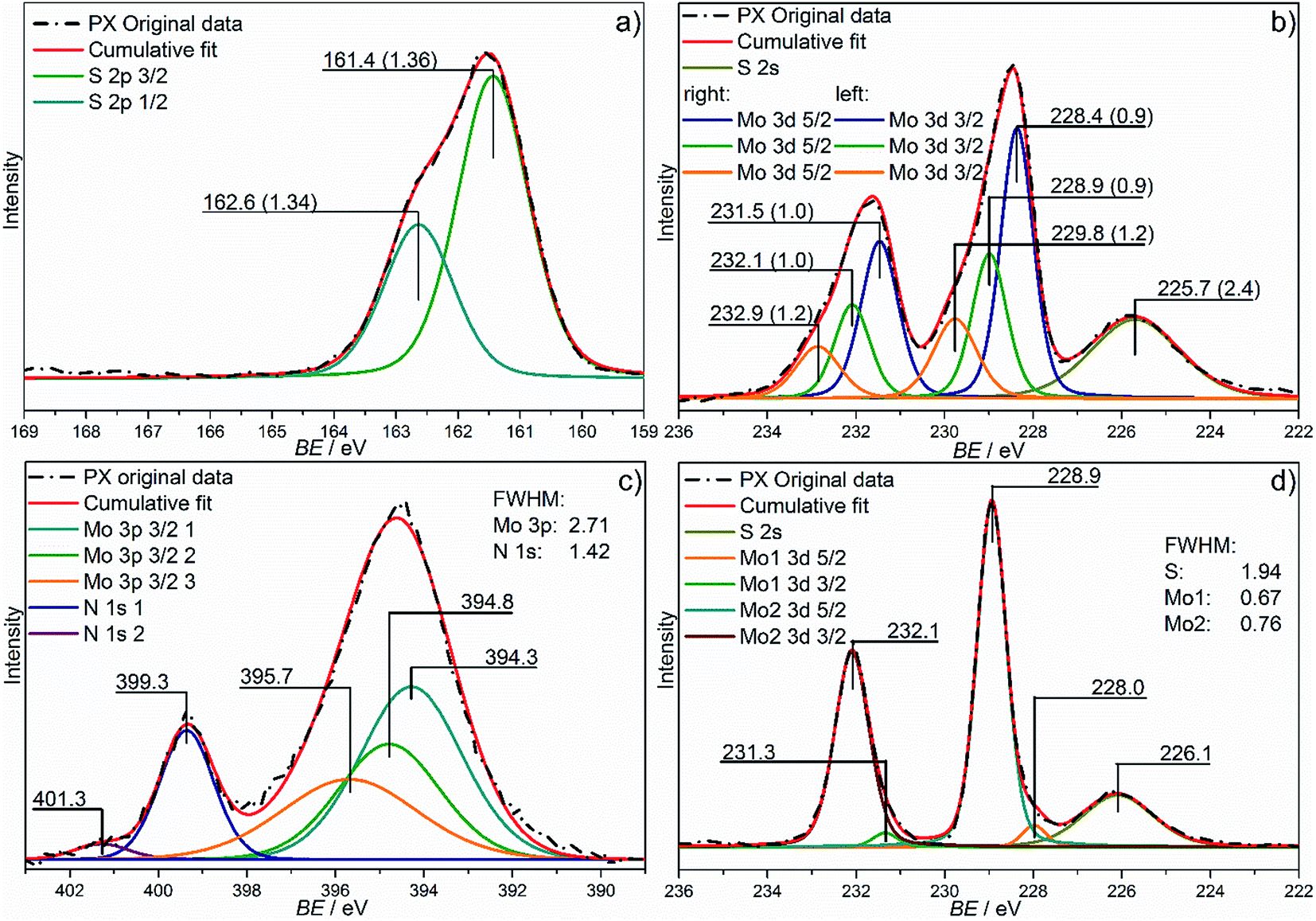 | ||
| Fig. 4 XPS measurements of the S 2p (a), Mo 3d (as obtained: (b) and heated to 450 °C: (d)), and the N 1s (c) region of PX. The dash-dotted lines represent the original data and the red lines the cumulative fits. | ||
Fig. 4a depicts the XPS spectrum of the S 2p region. The S 2p peak of PX can be fitted with contributions from S 2p3/2 and 2p1/2 levels with Eb = 161.4 and 162.6 eV respectively with a spin–orbit splitting of 1.2 eV. The presence of amorphous MoS3 can be safely excluded, because the S 2p region of MoS3 consists of two doublets with Eb = 162.0 and 163.3 eV for the two S 2p3/2 signals due to the presence of bridging S22−, apical S2− ligands (higher energy doublet) and terminal S22− and/or S2− (lower energy doublet).46–48,59–62 Typical binding energies of the S 2p3/2 level in MoS2 extracted from the NIST database range between 161.5 and 163 eV,63 with most data at around 162.2 eV. The lower value for the S 2p signal in PX indicates different bond strength and/or a differing binding mode of the S atoms. Most likely PX contains μs-S2− species as observed in K5[Mo3S4(CN)9]·3KCN·4H2O, for which an Eb = 161.6 eV was reported.53
Fig. 4b shows the Mo 3d and S 2s region of PX. The deconvolution of the Mo 3d region yields three distinct Mo species, i.e. three Mo species in different chemical environments. The main contribution comes from the doublet located at 228.4 and 231.5 eV (3d5/2 and 3d3/2) while two further doublets are located at 228.9/232.1 and 229.7/233.0 eV for the 5/2 and 3/2 levels (ratios are 3:2).
For crystalline MoS2 the Mo 3d5/2 signal occurs at 229.4 eV, which is near the signal observed at 228.9 eV. Hence, this Mo species is most likely surrounded by only sulfur. The shift of the Eb relative to values reported for crystalline MoS2 is caused by a different bonding situation. The most intense Mo 3d5/2 peak represents a Mo center with higher electron density compared to the middle MoIV peak being assigned to Mo in a predominantly S2− environment. Both NH3 and N2H4 are electron donors reducing the positive charge on the Mo center leading to a shift to lower Eb. Such shift to lower Eb is also observed for 1 T-MoS2 with an Eb of the 3d5/2 shifted by ≈1 eV to a lower binding energy. The third peak at 229.8 eV represents a Mo species with less electron density compared to the other two species although the small energy shift does not allow assigning a significant higher oxidation state and one can safely assume that this Mo center has a different environment.
All the results indicate that Mo in PX is surrounded predominantly by four S atoms in a glass-like manner, with the three different Mo species being surrounded by different numbers of NH3/N2H4 neighbors. The Mo center with the lowest Eb for Mo 3d5/2 has the largest number of NH3/N2H4 in the coordination environment while that with the highest Eb is surrounded only by few donor molecules.
2.2. Heating of PX – crystallization
The thermal stability of as-prepared PX was investigated by heating the sample either in a furnace or by irradiation with the electronic beam of the TEM in situ. The XRD patterns of two samples heated in the furnace are shown in Fig. 1 for comparison. Apparently, the heating process initiated nucleation and crystallization of a layered material after heating at 450 °C. The absence of the (103) and (105) reflections clearly evidences a pronounced turbostratic disorder. Assigning the reflection at 14° θ to the (002) plane leads to an interlayer distance of 6.6 Å which is about 0.4 Å larger than in crystalline MoS2 and smaller than the interlayer distance of (002) planes determined via TEM (6.37 Å). Using the Scherrer formula a size of coherently scattering domains of 2.7 nm is obtained from the (002) reflection, giving a number of 4 stacked layers, which is consistent with the histogram obtained from in situ TEM heating (cf. Fig. S7†). The pronounced shift of the (002) reflection to a lower scattering angle compared to crystalline MoS2 is typical for layered materials with low stacking and/or the presence of residues between the layers (see also below). The background at low scattering angles indicates the presence of slabs with a lower stacking degree than 4. Heating at 900 °C yields a more crystalline sample with a size of coherently scattering domains of about 10.8 nm, i.e. about 17 MoS2 layers are stacked. Despite the high temperature of 900 °C the material still exhibits a pronounced disorder (stacking faults, turbostratic disorder) as can be seen from the shapes of the reflections. It is gratifying to note that the XRD results of the heating experiments are consistent with the results from the TEM analysis.Irradiating the sample with a 300 kV electron beam, the characteristic (002) single slabs of crystalline MoS2 (space group: P63/mmc) were formed, as demonstrated in the HRTEM micrographs in Fig. 5b and e. The length and stacking numbers of these slabs increased with extended exposure times, cf. result of long-term irradiation experiments of Fig. 2c and f. By closer inspection of the FFTs of Fig. 2e and f the formation of (011), (013), and (−120) lattice planes were also observable under irradiation.
 | ||
| Fig. 5 (a), (d), (g), and (j): Temperature resolved series of TEM bright-field images of the as prepared MoS2 during the in situ heating experiment. (b), (e), (h), and (k): The enlarged region from the same position (marked with a red box) of the sample showing the development of the (002) slabs as temperature increased. (c), (f), (i) and (l) are the corresponding inverse FFT (IFFT) showing more clearly the contrast from the (002) slabs. The measured (002) d-spacing is indicated. The IFFT is generated by applying a band pass mask to the FFT pattern of HRTEM micrograph with a frequency range from 2.35 nm−1 to 7.50 nm−1, and filters out all the noises outside this range. It means that, in real space, the filtered IFFT images exhibit only those MoS2 lattice planes with d-spacings from 0.267 nm to 0.85 nm. | ||
Furthermore, applying a heating stage for in situ observation and a lower dose of electrons also revealed an increase of crystallinity of the as prepared pristine MoS2. TEM overview bright field images (Fig. 5) from the same position of the sample are shown from 300 °C up to 500 °C. The corresponding inverse FFT images (Fig. 5c, f, i and l) exhibit more clearly the development of the (002) slabs of MoS2. At 300 °C the sample is mainly amorphous (cf.Fig. 2c), and at T = 350 °C the first indication of formation of the (002) slabs is detectable (Fig. 2f). The measured dimension (0.63 nm) of the individual slab is comparable to the theoretical value (0.615 nm) of the (002) plane of MoS2. At 450 °C and 500 °C, the length of the slabs is increasing and they are becoming more ordered as depicted in Fig. 5i and l, which is a sign of an increase of crystallinity of the sample upon heating.
Surprisingly, in contrast to the irradiation experiment, the stacking height of the (002) slabs, and even the length of the slabs formed during heating are more uniform between the regions examined in the sample, indicating an evolution of mostly the same sizes of the MoS2 grains when the as prepared material is heated (cf.Fig. 5c and j) and the size distribution histograms in Fig. S8.† The histogram for the in situ heating to 500 °C shows a stacking number of 4 layers for the (002) plane, and a basal plane length of 2 nm. These values are in good agreement with those of the sample heated to 450 °C and examined by XRD (see below). On the other hand, the size distribution obtained by in situ irradiation for 1 h is more broadened, with a stacking number of 5 and a longer basal plane length of 4–5 nm. The two contrary observations might be due to different crystallization mechanisms under electron irradiation and under heat treatment for nanosized materials.64 Electron irradiation generates highly localized heating in the sample particles, especially when the thermal conductivity of the specimen is relatively low, for example, monolayer MoS2 or amorphous MoS2, which is also presenting itself in the PX sample here. While in the thermal heating process, the good thermal conductivity of the carbon lacy network created a more uniform heat distribution over the whole sample grid, thus a more uniform size of MoS2 particles was produced.
The crystallization of MoS2 slabs can be monitored by Raman spectroscopy. The two typical modes E12g and A1g start to develop after heating PX at T = 450 °C and the intensity increased for the sample treated at T = 900 °C (see Fig. S1†). The highest intensity is observed for commercial MoS2 which is characterized by the largest number of stacked MoS2 slabs. The energy difference between the two Raman modes of MoS2 may be used to estimate the number of stacked layers. Four stacked layers showed a difference of the Raman modes of about 24.3 cm−1, which fits perfectly for the sample heated at 450 °C and is in full agreement with the result of the XRD investigation, where also four slabs were estimated. The larger energy separation obtained for PX900 and bulk MoS2 is in line with the larger number of stacked MoS2 slabs.46
Upon heating to 450 °C the compound crystallizes at least partially to form MoS2 slabs and on the first sight the XPS spectrum of the Mo 3d region (Fig. 4d) is very similar to that of MoS2. The spectrum of the N 1s region only shows the Mo 3p peaks while the N 1s peaks have vanished. In the Mo 3d spectrum, there seems to be a small asymmetry of the peaks towards lower binding energies, which has been fitted as a second Mo species at 228.1 and 231.2 eV. However, due to uncertainties inherent in Shirley background subtraction this signal should be treated with caution. The predominant Mo species appears at 228.9 eV and 232.1 eV (Mo 3d5/2 and 3d3/2). In contrast to the as prepared PX material only two Mo species can be identified, one of which is hardly significant. This observation is in good agreement with formation of crystalline MoS2 slabs (minor component) embedded in a matrix of glassy like amorphous molybdenum sulfide (dominating component), a scenario which would not necessarily require two different Mo 3d peaks as both species are close to be MoIV. The higher binding energy species observed in the spectrum of as prepared PX disappeared, which is accompanied by the disappearance of the N 1s signal (Fig. S7†). Furthermore the main Mo peak of the heated sample has the same binding energy as the second species in PX as prepared. These observations substantiate that the other Mo species detected in the as prepared material were associated with the presence of nitrogen containing ligands in the sample.
2.3. Visible light driven hydrogen evolution
Many publications in literature deal with the application of MoS2/MoS3 based materials as catalysts for the light driven photocatalytic hydrogen production.60,65–71 Commonly used multicomponent catalyst systems for this purpose contain a photosensitizer (PS) for light harvesting, a sacrificial agent (SA) as electron donor and a proton source, usually water, besides a water reduction catalyst (WRC). For solubility or performance reasons organic solvents are added. The results presented herein were obtained from multicomponent catalyst systems with [Ru(bpy)3](PF6)2 as PS, water as proton source and triethylamine as SA in acetonitrile. All materials (PX, PX450, PX900) as well as two additional reference samples (commercial MoS2 and a sample of MoS2 prepared from ATM at 350 °C) were tested as WRCs in this system and proved active in light driven proton reduction (Fig. 6). Although activity of all catalyst systems ceased after ∼20 h of irradiation with visible light, this is most certainly due to degradation of the PS [Ru(bpy)3](PF6)2 as reported previously.72 This was additionally demonstrated by reapplication of PS after the reaction had stopped completely. After equilibration the reaction was reinitialized and showed the same hydrogen evolution trend as before, indicating that the PS is degraded during the experiment while the catalyst PX is stable under the reaction conditions (Fig. S11†). Furthermore, the chemical analysis show no major difference in composition to the pristine material and the XRD pattern of the post-catalytic material (Fig. 1, green trace) evidence no significant change. Because the catalyst was washed three times with isopropyl alcohol the carbon and hydrogen contents are increased, but the ratios of Mo:S and Mo:N did not change. A follow-up catalytic test with the recovered material exhibited significant catalytic activity even though a solid comparison is not possible due to a high loss of material during the recovery process, since only 10 mg were used for the respective initial catalytic tests.
 | ||
| Fig. 6 Comparison of activities (t = 1 h) of different catalysts based on MoS2 in photocatalytic hydrogen evolution from mixtures containing triethylamine (8 mL), water (3 mL) and [Ru(bpy)3](PF6)2 (1 mM) in acetonitrile (10 mL). PX as obtained and heated samples are displayed. For comparison results with MoS2 prepared from ATM by decomposition at 350 °C and commercial bulk MoS2 are displayed. | ||
Of all materials tested, PX showed the best results with a hydrogen production of 14.7 μmol mg−1 catalyst in 1 h (Fig. 6). This remarkable activity decreased only slightly after heating a sample of PX to 150 °C to exclude the contribution of gaseous species (Fig. 6). Heating of the catalyst material to temperatures above the crystallization point however caused a considerable decrease in activity compared to PX with values of 5.17 μmol mg−1 catalyst and 4.25 μmol mg−1 catalyst in 1 h for heat treatment at 450 °C and 900 °C, respectively.
This is still a clear enhancement of activity compared to a sample of commercial bulk MoS2, which produced 1.2 μmol mg−1 catalyst in 1 h in an identical multi-component catalyst system. In accordance with previous results this suggests enhanced activity in catalytic proton reduction with decreasing particle size for MoS2 materials.73 Further enhancement of photocatalytic activity for amorphous samples compared to their crystalline counterparts has not been reported yet. Further support that the amorphous state improves the properties of MoS2 materials as WRCs in light driven proton reduction arises from a comparison to a conventional nanocrystalline sample of MoS2. This was obtained by thermal decomposition of ATM at 350 °C and shows similar properties to PX450 with respect to composition as well as structure and stacking behavior.72,74 Even activity in light driven proton reduction in an identical multi-component catalyst system is similar to PX450, thus suggesting the electronic properties and local environment of PX as reason for the remarkable activity of the material. The present results support reports21–23 that amorphous Mo sulfide based materials are partially more active than crystalline MoS2. With the MoS2 materials presented here, a direct comparison of amorphous to crystalline catalysts is available, which results in the conclusion that amorphous MoS2 can be basis for highly active WRCs, too. Concerning the catalytically active sites in PX a direct comparison with models widely accepted for crystalline MoS2 cannot be done. It was demonstrated that in crystalline MoS2 the so-called Mo-edge is catalytically active for hydrogen evolution while the S-edge seems to be catalytically inactive.74,75 Because PX is X-ray amorphous no such edges exist and only for the crystalline samples investigated here the model can be invoked to explain the catalytic properties.
3. Conclusion
In conclusion a new preparation method utilizing kinetic control was developed to synthesize an amorphous molybdenum sulfide based material and the product has been thoroughly characterized with various methods to elucidate the chemical nature of the constituents. The product is completely amorphous as evidenced by XRD, TEM and Raman spectroscopy. According to chemical analysis the Mo:S ratio is near 1:2 like in crystalline 2H–MoS2 excluding the formation of amorphous MoS3. Appreciable amounts of N are present and taking into account the XPS, NMR and spectroscopic data different locally varying environments of the Mo centers must be postulated. The material shows an unusual thermal stability and only if the samples is heated at T ≥ 350 °C crystallization takes place being accompanied by a mass loss, which is primarily attributed to the emission of hydrazine leading to formation of 2H–MoS2. The crystallization process can be controlled by applying different temperatures or dwelling times or even using different methods like the electron beam of a TEM. The photocatalytic activity is highest for the amorphous material indicating that most likely Mo centers with N and S as neighbors are the catalytic active species.
4. Experimental section
4.1. Sample preparation
The starting materials (ammonium heptamolybdate and ammonia) were obtained in high-purity from Sigma-Aldrich. Adding ammonium heptamolybdate ((NH4)6Mo7O24·4H2O, 30 g) to a 25 wt% solution of ammonia (200 mL) and bubbling H2S for half an hour through the solution leads to crystallization of the precursor ammonium tetrathiomolybdate ((NH4)2MoS4, ATM) which was obtained in high yield.The amorphous molybdenum sulfide based material (PX) has been synthesized by storing freshly prepared ATM in a desiccator over hydrazine monohydrate (99%) (Caution! Highly toxic material!). Typically hydrazine (20 mL) was deposited in a Petri dish located in the lower chamber of a desiccator and after ball milling ATM (1 h, 2 g) was finely dispersed on another Petri dish and stored above. The reaction at room temperature is observed by change in color of ATM to anthracite and is completed in about twenty to thirty days as evidenced by the formation of insoluble products. The complete conversion of ATM was further confirmed as the product (PX) did not give any red color on reaction with water. However, products isolated before a period of 20–30 d always contained still some ATM as evidenced by a red coloration on treatment with water. The gas–solid reaction can be significantly accelerated increasing the temperature. At about 32 °C the reaction is completed after 7 d. After completion of the reaction the Petri dish was taken out and kept in another desiccator containing silica granules as desiccant for yet another day for thorough drying after which the reaction product was weighed.
4.2. Characterization
X-Ray powder patterns were recorded with a STOE STADI-P instrument (monochromatized CuKα1 radiation, λ = 1.54056 Å) in transmission mode with a position sensitive detector (Mythen).4.3. Raman
Raman measurements were carried out on a Dilor RamanXY instrument equipped with an Ar/Kr mixed gas ion laser (type: RM2018 by Spectra-Physics) with various possible excitation wavelengths (488–648.2 nm) and a N2-cooled CCD detector for imaging. The sample was rotated during the measurement and cooled to 20 K.4.4. Thermal analysis
DTA-TG was performed in a nitrogen atmosphere (purity: 5.0; heating rate 4 K min−1; flow rate: 75 mL min−1; Al2O3 crucibles) using a Netzsch STA-409CD instrument.4.5. Spectroscopy
IR/NIR/FIR spectra were recorded at room temperature using an Alpha P FT-IR spectrometer from Bruker. The IR spectra were measured from 200 to 4000 cm−1.4.6. Transmission electron microscopy
TEM investigations were performed with a Tecnai 30 STwin microscope (300 kV, FEG cathode, Cs = 1.2 mm). The samples for TEM investigations were prepared as follows. A small amount of precursor powder was suspended in methanol and ultrasonically grinded for 15 min. Afterwards, a transfer of the suspended powder on a TEM support (a lacey carbon film on 200 mesh cooper grid) was carried out by dipping the TEM support into the solution. The prepared TEM samples were dried at room temperature for several minutes. All images were recorded with a Gatan Multiscan CCD camera (2k × 2k) and evaluated (including Fourier analyses) with the program Digital Micrograph (Gatan). EDS analyses were performed in the TEM mode with a Si/Li detector (EDAX System).4.7. X-ray photoelectron spectroscopy
XPS measurements were carried out in an ultra-high vacuum (UHV) chamber equipped with a monochromatic Al Kα X-ray source (hν = 1486.6 eV), operated at 14.5 kV and 35 mA, and a high resolution Gammadata-Scienta SES 2002 analyzer. The base pressure in the measurement chamber was maintained at about 5 × 10−10 mbar. The measurements were done in the fixed transmission mode with pass energy of 200 eV resulting in an overall energy resolution of 0.25 eV. A flood gun was applied to compensate charging effects. High-resolution spectra for C 1s, O 1s, N 1s, S 2p and Mo 3d photoelectron lines were recorded. The C 1s signal (284.5 eV) was used as an internal standard for calibration of the spectra. The Casa XPS software with Gaussian–Lorentzian product function and Shirley background subtraction was used for peak deconvolution, whereas the ratio of the Lorentzian to the Gaussian portion of the function was set to 30%/70% (profile shape factor μ = 0.3). The energy scale calibration of the machine was carried out using Au 4f7/2 = 84.0 eV, Ag 3d3/2 = 368.3 eV and Cu 2p3/2 = 932.7 eV.4.8. 1H MAS NMR
NMR spectra were recorded at ambient temperature on an Avance III HD 600 solid-state NMR spectrometer (Bruker) with an external magnetic field 14.1 T, operating at a frequency of 600.1 MHz using a spin echo pulse sequence. The sample was contained in a 1.3 mm ZrO2 rotor which was mounted in a standard double resonance MAS probe (Bruker) and spun at 60 kHz. The spectrum was referenced relative to TMS.4.9. 1H15N cross-polarization (CP) MAS NMR
NMR spectra were recorded at ambient temperature on an Avance III HD 400 solid-state NMR spectrometer (Bruker) with an external magnetic field of 9.4 T, operating at frequencies of 400.1 MHz and 40.6 MHz for 1H and 15N, respectively. The sample was contained in a 4 mm ZrO2 rotor which was mounted in a standard triple resonance MAS probe (Bruker) and spun at 10 kHz. During a contact time of 1 ms a ramped-amplitude (RAMP) CP pulse, the RF field was linearly varied about 30% on 1H, was used. Due to the metal-like properties of the sample severe heating and detuning of the probe took place upon RF irradiation. Therefore low power continuous wave (CW) decoupling (roughly 10 kHz RF field) was applied during the acquisition of the 15N signal. The recycle delay was set to 1s. The 15N chemical shifts were referenced relative to nitromethane.4.10. Photocatalytic reactions
All reactions were carried out in a double-walled thermostatically controlled reaction vessel using an automatically operating burette (MesSen Nord GmbH, Stäbelow, Germany) described previously.76 All liquids were degassed by ultrasound treatment for at least 15 min prior to use. Triethylamine and acetonitrile were dried on CaH2 and distilled under argon. A 300 W Xe lamp was used as light source along with a 420 nm cut-off filter, which leaves a light power at the flask of about 900 mW. In a standard procedure 10 mL of a 10−3 M solution of [Ru(bpy)3](PF6)2 in acetonitrile, 8 mL trimethylamine and 3 mL distilled water were transferred via syringe into an air- and moisture-free glass reaction vessel which contained 10 mg of the WRC. After equilibration the freshly assembled multicomponent catalyst system was irradiated with visible light at isothermal conditions for both liquid and gaseous phase at 298.15 K. At the end of the reaction a gas sample was taken and analyzed by GC.Acknowledgements
L. Dura and T. Beweries thank Dr Sven Hansen (LIKAT) for assistance and Prof. Uwe Rosenthal (LIKAT) for support and fruitful discussions.References
- J. D. Benck, T. R. Hellstern, J. Kibsgaard, P. Chakthranont and T. F. Jaramillo, ACS Catal., 2014, 4, 3957–3971 CrossRef CAS.
- C. G. Morales-Guio and X. Hu, Acc. Chem. Res., 2014, 47, 2671–2681 CrossRef CAS PubMed.
- X. Zong, G. P. Wu, H. Yan, G. Ma, J. Shi, F. Wen, L. Wang and C. Li, J. Phys. Chem. C, 2010, 114(4), 1963–1968 CAS.
- W. K. Ho, J. C. Yu, J. Lin, J. Yu and P. Li, Langmuir, 2004, 20(14), 5865 CrossRef CAS PubMed.
- D. Merki and X. Hu, Energy Environ. Sci., 2011, 4(10), 3878 CAS.
- H. Tributsch, Z. Naturforsch. A, 1977, 32(9), 972 Search PubMed.
- H. J. Tributsch and C. Bennett, J. Electroanal. Chem., 1977, 81, 97 CrossRef CAS.
- H. Vrubel and X. Hu, ACS Catal., 2013, 3, 2002 CrossRef CAS.
- X. Ge, L. Chen, L. Zhang, Y. Wen, A. Hirata and M. Chen, Adv. Mater., 2014, 26, 3100 CrossRef CAS PubMed.
- D. Wang, Z. Pan, Z. Wu, Z. Wang and Z. Liu, J. Power Sources, 2014, 264, 229 CrossRef CAS.
- B. Hinnemann, P. G. Moses, J. Bonde, K. P. Jørgensen, J. H. Nielsen, S. Horch, I. Chorkendorff and J. K. Nørskov, J. Am. Chem. Soc., 2005, 127, 5308–5309 CrossRef CAS PubMed.
- T. F. Jaramillo, K. P. Jørgensen, J. Bonde, J. J. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100–102 CrossRef CAS PubMed.
- H. Y. Li, L. Wang, Y. Xie, G. Liang, H. Hong and H. Dai, J. Am. Chem. Soc., 2011, 133, 7296–7299 CrossRef PubMed.
- H. Wang, Z. Lu, S. Xu, D. Kong, J. J. Cha, G. Zheng, P.-C. Hsu, K. Yan, D. Bradshaw, F. B. Prinz and Y. Cui, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 19701–19706 CrossRef CAS PubMed.
- C. G. Morales-Guio, L.-A. Stern and X. Hu, Chem. Soc. Rev., 2014, 43, 6555 RSC.
- A. B. Laursen, S. Kegnaes, S. Dahl and I. Chorkendorff, Energy Environ. Sci., 2012, 5, 5577–5591 CAS.
- D. Merki and X. Hu, Energy Environ. Sci., 2011, 4, 3878–3888 CAS.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253 RSC.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503 CrossRef CAS PubMed.
- F. E. Osterloh, Chem. Mater., 2008, 20, 35 CrossRef CAS.
- D. Y. C. Leung, X. Fu, C. Wang, M. Ni, M. K. H. Leung, X. Wang and X. Fu, ChemSusChem, 2010, 3, 681 CrossRef CAS PubMed.
- C. G. Morales-Guio and X. Hu, Acc. Chem. Res., 2014, 47, 2671–2681 CrossRef CAS PubMed.
- D. Merki, S. Fierro, H. Vrubel and X. Hu, Chem. Sci., 2011, 2, 1262–1267 RSC.
- H. Vrubel, D. Merki and X. Hu, Energy Environ. Sci., 2012, 5, 6136–6144 CAS.
- T.-W. Lin, C.-J. Liu and J.-Y. Lin, Appl. Catal., B, 2013, 134–135, 75–82 CrossRef CAS.
- Y. Li, Y. Yu, Y. Huang, R. A. Nielsen, W. A. Goddard, Y. Li and L. Cao, ACS Catal., 2015, 5, 448–455 CrossRef CAS.
- J. D. Benck, Z. Chen, L. Y. Kuritzky, A. J. Forman and T. F. Jaramillo, ACS Catal., 2012, 2, 1916–1923 CrossRef CAS.
- C.-L. Hsu, Y.-H. Chang, T.-Y. Chen, C.-C. Tseng, K.-H. Wei and L.-J. Li, Int. J. Hydrogen Energy, 2014, 39, 4788–4793 CrossRef CAS.
- Y.-H. Chang, C.-T. Lin, T.-Y. Chen, C.-L. Hsu, Y.-H. Lee, W. Zhang, K.-H. Wei and L.-J. Li, Adv. Mater., 2013, 25, 756–760 CrossRef CAS PubMed.
- Y.-H. Chang, F.-Y. Wu, T.-Y. Chen, C.-L. Hsu, C.-H. Chen, F. Wiryo, K.-H. Wei, C.-Y. Chiang and L.-J. Li, Small, 2014, 10, 895–900 CrossRef CAS PubMed.
- S. J. Hibble and G. B. Wood, J. Am. Chem. Soc., 2004, 126, 959–965 CrossRef CAS PubMed.
- S. J. Hibble, R. I. Walton, D. M. Pickup and A. C. Hannon, J. Non-Cryst. Solids, 1998, 232–234, 434–439 CrossRef CAS.
- T. Weber, J. C. Muijsers and J. W. Niemantsverdriet, J. Phys. Chem., 1995, 99, 9194–9200 CrossRef CAS.
- S. J. Hibble, D. A. Rice, D. M. Pickup and M. P. Beer, Inorg. Chem., 1995, 34, 5109–5113 CrossRef CAS.
- J. D. Benck, Z. B. Chen, L. Y. Kuritzky, A. J. Forman and T. F. Jaramillo, ACS Catal., 2012, 2, 1916–1923 CrossRef CAS.
- D. Merki, S. Fierro, H. Vrubel and X. L. Hu, Chem. Sci., 2011, 2, 1262–1267 RSC.
- D. Merki, H. Vrubel, L. Rovelli, S. Fierro and X. Hu, Chem. Sci., 2012, 3, 2515–2525 RSC.
- P. Afanasiev, G.-F. Xia, G. Berhault, B. Jouguet and M. Lacroix, Chem. Mater., 1999, 11, 3216–3219 CrossRef CAS.
- A. Müller, V. Fedin, K. Hegetschweiler and W. Amrein, Chem. Commun., 1992, 24, 1795–1796 RSC.
- A. Müller, E. Diemann, E. Krickmeyer, H.-J. Walberg, H. Bögge and A. Armatage, Eur. J. Solid State Inorg. Chem., 1993, 30, 565 Search PubMed.
- H. Jiao, Y. Li, B. Delmon and J. Halet, J. Am. Chem. Soc., 2001, 123, 7334 CrossRef CAS PubMed.
- S. J. Hibble and G. B. Wood, J. Am. Chem. Soc., 2004, 126, 959–965 CrossRef CAS PubMed.
- I. Bezverkhy, P. Afanasiev and M. Lacroix, Inorg. Chem., 2000, 39, 5416–5417 CrossRef CAS PubMed.
- P. Afanasiev, G.-F. Xia, G. Berhault, B. Jouguet and M. Lacroix, Chem. Mater., 1999, 11, 3216–3219 CrossRef CAS.
- I. Bezverkhyy, P. l. Afanasiev and M. l Lacroix, J. Catal., 2005, 230, 133–139 CrossRef CAS.
- P. Joensen, E. D. Crozier, N. Alberding and R. F. Frindt, J. Phys. C: Solid State Phys., 1987, 20, 4043 CrossRef CAS.
- C. Lee, H. Yan, L. E. Brus, T. F. Heinz, J. Hone and S. Ryu, ACS Nano, 2010, 4, 2695 CrossRef CAS PubMed.
- A. Molina-Sánchez and L. Wirtz, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 155413 CrossRef.
- T. Weber, J. C. Muijsers, J. H. M. C. van Wolput, C. P. J. Verhagen and J. W. Niemantsverdriet, J. Phys. Chem., 1996, 100, 14144 CrossRef CAS.
- M. Witanowski, L. Stefaniak and G. A. Webb, Annu. Rep. NMR Spectrosc., 1987, 18, 1 CrossRef.
- L. D. Field, H. L. Li and S. J. Dalgarno, Inorg. Chem., 2010, 49, 6214 CrossRef CAS PubMed.
- F. Haase and J. Sauer, J. Phys. Chem., 1994, 98, 3083 CrossRef CAS.
- G. V. Lagodzinskaya, N. G. Yunda and G. B. Manelis, Russ. Chem. Bull., Int. Ed., 2003, 55, 577 Search PubMed.
- G. Albertin, S. Antoniutti and C. Girardi, Polyhedron, 2012, 38, 162 CrossRef CAS.
- G. Albertin, S. Antoniutti, A. Bacchi, M. Bergamo, E. Bordignon and G. Pelizzi, Inorg. Chem., 1998, 37, 479 CrossRef CAS PubMed.
- K. J. D. Mackenzie and M. E. Smith, Multinuclear Solid-state NMR of Inorganic Materials, Pergamon Materials Series Volume 6, Elsevier Science Ltd, Oxford, 2002 Search PubMed.
- D. N. Hendrickson, J. M. Hollander and W. L. Jolly, Inorg. Chem., 1969, 8, 2642 CrossRef CAS.
- K. Artyushkova, B. Kiefer, B. Halevi, A. Knop-Gericke, R. Schlögl and P. Atanassov, Chem. Commun., 2013, 49, 2539 RSC.
- T. Weber, J. C. Muijsers and J. W. Niemantsverdriet, J. Phys. Chem., 1995, 99, 9194 CrossRef CAS.
- T. A. Patterson, J. C. Carber, D. E. Leyden and D. M. Hercules, J. Phys. Chem., 1976, 80(15), 1700 CrossRef CAS.
- D. Merki, S. Fierro, H. Vrubel and X. Hu, Chem. Sci., 2011, 2, 1262 RSC.
- H. Vrubel, D. Merki and X. Hu, Energy Environ. Sci., 2012, 5, 6136 CAS.
- NIST X-ray Photoelectron Spectroscopy Database, Version 4.1, National Institute of Standards and Technology, Gaithersburg, MD 20899-8370, 2015, http://srdata.nist.gov/xps/, accessed: January Search PubMed.
- M. Deng, V. Hrkac, L. Kienleet al., to be submitted.
- J. Djamil, S. A. Segler, A. Dabrowski, W. Bensch, A. Lotnyk, U. Schürmann, L. Kienle, S. Hansen and T. Beweries, Dalton Trans., 2013, 42, 1287 RSC.
- X. Zong, Y. Na, F. Y. Wen, G. J. Ma, J. H. Yang, D. G. Wang, Y. Ma, W. Wang, L. Sun and C. Li, Chem. Commun., 2009, 30, 4536 RSC.
- X. Zong, G. P. Wu, H. J. Yan, G. J. Ma, J. Y. Shi, F. Y. Wen, L. Wang and C. Li, J. Phys. Chem. C, 2010, 114, 1963 CAS.
- F. A. Frame and F. E. Osterloh, J. Phys. Chem. C, 2010, 114, 10628 CAS.
- A. Sobczynski, J. Catal., 1991, 131, 156 CrossRef CAS.
- S. Kanda, T. Akita, M. Fujishima and H. Tada, J. Colloid Interface Sci., 2011, 354, 607 CrossRef CAS PubMed.
- Q. J. Xiang, J. G. Yu and M. Jaroniec, J. Am. Chem. Soc., 2012, 134, 6575 CrossRef CAS PubMed.
- A. Vaidyalingam and P. K. Dutta, Anal. Chem., 2000, 72, 5219 CrossRef CAS PubMed.
- T. F. Jaramillo, K. P. Jörgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 319, 100–102 CrossRef PubMed.
- H. G. Sanchez Casalongue, J. D. Benck, C. Tsai, R. K. B. Karlsson, S. Kaya, M. L. Ng, L. G. M. Pettersson, F. Abild-Pedersen, J. K. Nørskov, H. Ogasawara, T. F. Jaramillo and A. Nilsson, J. Phys. Chem. C, 2014, 118(50), 29252 Search PubMed.
- H. Wang, C. Tsai, D. Kong, K. Chan, F. Abild-Pedersen, J. K. Nørskov and Y. Cui, Nano Res., 2015, 8, 566–575 CrossRef CAS.
- T. Beweries, J. Thomas, M. Klahn, A. Schulz, D. Heller and U. Rosenthal, ChemCatChem, 2011, 3, 1865–1868 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra14438h |
| This journal is © The Royal Society of Chemistry 2015 |