
Emulsion-templated poly(acrylamide)s by using polyvinyl alcohol (PVA) stabilized CO2-in-water emulsions and their applications in tissue engineering scaffolds
Wei Luoa,
Ran Xub,
Yunfei Liua,
Irshad Hussainc,
Qunwei Lu*b and
Bien Tan*a
aKey Laboratory for Large-Format Battery Materials and System, Ministry of Education, School of Chemistry and Chemical Engineering, Huazhong University of Science and Technology, Wuhan 430074, PR China. E-mail: bien.tan@mail.hust.edu.cn; Fax: +86-027-87543632; Tel: +86-027-87558172
bKey Laboratory of Molecular Biophysics of Ministry of Education, School of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, China. E-mail: luqw@hust.edu.cn
cDepartment of Chemistry, SBA School of Science & Engineering, Lahore University of Management Sciences (LUMS), D.H.A., Lahore Cantt, 54792, Pakistan
First published on 16th October 2015
Abstract
The key challenge in the formation of stable CO2-in-water (C/W) emulsions has always been the availability of effective surfactants. Most of the surfactants being used for this purpose are expensive and difficult to synthesize, rendering the formation of C/W emulsions an uneconomical and unfavorable process. In this work, we report the use of commercial polyvinyl alcohol as an effective stabilizer for the formation of C/W emulsions at low temperatures. Porous emulsion-templated materials were prepared by the polymerization of the continuous phase of C/W emulsions. The open-cell morphology of the emulsion-templated materials was observed by scanning electron microscopy. To tune the morphology of the porous structures, the influence of the stabilizer concentration and the process of polymerization were investigated. These porous PAM materials were further evaluated for cellular growth and proliferation to demonstrate their applications in tissue engineering.
Introduction
Templating of high internal phase emulsions (HIPEs) provides an efficient approach to synthesize hierarchically porous materials with a range of functionalities and applications. In high internal phase emulsions, the volume of the dispersed phase is higher than 74% of the total emulsion volume that is equivalent to the maximum packing fraction of monodisperse hard spheres.1 Polymerized high internal phase emulsions (polyHIPEs), with a highly porous and interconnected morphology, are materials prepared by polymerizing the continuous phase of HIPE. The cavity size of polyHIPEs is easily tunable by controlling the factors such as surfactant type,2 its concentration3 and dispersed phase volume to mention a few.4 And poly(HIPEs) have been applied for many applications including enzyme immobilization and separation,5 hydrogen or CO2 storage,6,7 in vitro release,8 photocatalysis,9 oil spill reclamation and wastewater treatment10,11 etc.However, in the traditional HIPE polymerization, a large amount of organic solvent, which is usually difficult to remove, is an indispensable factor and is thus associated with serious environmental concerns. This problem has partly been resolved by templating supercritical CO2-in-water (C/W) emulsions. This technique allows the synthesis of materials with well-defined porous structures without the use of any volatile organic solvents, but only water and CO2 as the dispersion medium. Moreover, CO2 is nontoxic, non-flammable, and naturally abundant12 alternative to the organic solvents, which can be readily removed from a polyHIPE scaffold by depressurization of the system.13
Butler et al. used perfluoropolyether (PFPE) as surfactant and poly(vinyl alcohol) (PVA) as cosurfactant to stabilize C/W emulsions of acrylamide polymers.13,14 Partap et al. used ammonium perfluoropolyether (PFPE–NH4) as surfactant to produce emulsion templated alginate hydrogels.15 Bing et al. successfully prepared porous CaCO3/PAM composites by C/W emulsion templating method by stabilizing the HIPE with a fluorosurfactant FC4430, PVA and CaCO3 particles.16 Palocci et al. reported the preparation of dextran-stabilized polyHIPEs in the presence of ammonium perfluoropolyether (PFPE–NH4), using supercritical CO2 as the internal phase.17 Our group has reported the synthesis of PVA–PEG-based di- and tri-block surfactants, which were found to stabilize C/W HIPEs.18–20 We also reported the use of the combination of reversible addition–fragmentation chain transfer radical polymerization (RAFT) and atom transfer radical polymerization (ATRP) methods to synthesize PVAc-b-PDMAEMA amphiphilic copolymers, which were also found to be effective surfactants to prepare stable CO2-in-water emulsions and PAM monolithic materials.21
However, CO2 is relatively a poor solvent for the majority of materials. It is widely accepted that the key challenge in C/W emulsion polymerization is the design of the surfactant.22 In the last several years, a rapid progress in the design and synthesis of CO2-philic hydrocarbon-based polymers has been witnessed. Howdle et al. synthesized CO2-soluble vinyl pivalate hydrocarbon stabilisers and poly(vinyl acetate-alt-dibutyl maleate) (PVAc-alt-PDBM) copolymer via RAFT polymerization.23,24 Vinyl ester copolymers poly(vinyl butyrate) (PVBu) and poly(vinyl octanoate) (PVOc) have also been designed/synthesized for their use as stabilisers for dispersion polymerisation in scCO2.25 In this regard, we reported the synthesis of oligomer vinyl acetate with different topologies and functional oligo(vinyl acetate) bearing bipyridine moieties by RAFT/MADIX method.26,27 Johnston et al. developed the nonionic hydrocarbon surfactants and investigated the morphology and stability of CO2-in-water foams.28 However, the high cost and difficulty of complex synthetic and purification processes usually render the formation of C/W emulsions uneconomical and an unfavorable process.
The commercial partially hydrolyzed PVA contains significant amount of poly(vinyl acetate) (PVAc) which is known to have extraordinarily high solubility in CO2 compared to other vinyl hydrocarbon polymers.29,30 Therefore, the PVA (88% or 80% hydrolyzed) can show amphiphilicity in the CO2/H2O system, which may stabilize CO2/H2O emulsions. Moreover, compared with the traditional surfactants, PVA is more suitable for biological applications because of its biocompatibility. Butler et al. and Chen et al. reported that PVA can further stabilize the emulsion system, the role of PVA being only to change the viscosity of C/W emulsions as a cosurfactant.13,14,20 However, no stable emulsions were formed by using PVA only as a stabilizer.
Recently,31 we successfully prepared stable C/W emulsions by using the commercial partially hydrolyzed PVA as stabilizer without additional surfactants, and PVA hydrogels, which can be used as tissue engineering scaffolds. PVA solution has higher viscosity, which helps to increase the stability of emulsions. By decreasing the temperature, the solubility of CO2 in water was increased32 and concentrated C/W emulsions became even more stable.14 Therefore, CO2/H2O emulsions can easily be formed using partially hydrolyzed PVA at low temperature. It was observed that human fibroblast cells can grow on the surfaces and also penetrate into the pores of PVA hydrogels.
In this work, we further use the commercial partially hydrolyzed PVA to stabilize AM containing C/W emulsions, which are not easy to get stabilized even with traditional surfactants, to prepare porous emulsion-templated PAM monoliths. The open-cell morphology of the emulsion-templated materials was confirmed by scanning electron microscopic analysis. The influence of stabilizer concentration and the process of polymerization on the preparation of C/W emulsion-templated PAM-based materials were also investigated in more detail. At last, the monolithic materials were used as tissue engineering scaffolds to guide cell growth, and the results indicate that the H9c2 cardiac muscle cells can grow and proliferate well on the surface of the materials as observed by fluorescence assay.
Experimental
Materials
PVA (88% hydrolyzed, 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 and 88000 g mol−1), ethanol and formaldehyde solution (≥36.0% in H2O) were purchased from Aladdin. PVA (80% hydrolyzed, 10000 g mol−1) was purchased from Sigma-Aldrich. Potassium persulfate (KPS, Sinopharm, 99%) was recrystallized twice from water. Acrylamide (AM, 98%), N, N′-methylene bis(acrylamide) (MBAM, 98%), and tetramethylethylenediamine (98%) were all purchased from Sinopharm and used as received. H9c2 cardiac muscle cells were obtained from the American type culture collection (ATCC). Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% v/v fetal bovine serum (FBS) were purchased from GIBCO. 4′,6-Diamidino-2-phenylindole (DAPI) was obtained from Beyotime. High purity carbon dioxide (99.99%) was purchased from Ming Hui Gas.
000 g mol−1 and 88000 g mol−1), ethanol and formaldehyde solution (≥36.0% in H2O) were purchased from Aladdin. PVA (80% hydrolyzed, 10000 g mol−1) was purchased from Sigma-Aldrich. Potassium persulfate (KPS, Sinopharm, 99%) was recrystallized twice from water. Acrylamide (AM, 98%), N, N′-methylene bis(acrylamide) (MBAM, 98%), and tetramethylethylenediamine (98%) were all purchased from Sinopharm and used as received. H9c2 cardiac muscle cells were obtained from the American type culture collection (ATCC). Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% v/v fetal bovine serum (FBS) were purchased from GIBCO. 4′,6-Diamidino-2-phenylindole (DAPI) was obtained from Beyotime. High purity carbon dioxide (99.99%) was purchased from Ming Hui Gas.
Formation of stable C/W HIPEs
Homogeneous PVA solution was prepared by dissolving PVA in hot water (about 80 °C) and then cooled down to room temperature. Then aqueous solutions (2 mL) of monomers (40% w/v in H2O), K2S2O8 initiator (2% w/v based on monomers) and PVA were added in a 10 cm3 high-pressure autoclave, with visual stainless steel window, purged by a slow flow of CO2 gas for 5 min to ensure the replacement of any oxygen with CO2. After that, high purity CO2 gas was continuously pumped into the reactor by a high-pressure pump up to the desired pressure (room temperature, vessel pressure ≈ 100 bar) under vigorous magnetic stirring (1000 rpm). Ice cubes were used to cover the reaction vessel to lower the temperature to about 8 °C. Milky-white C/W emulsions were formed in the reaction vessel by increasing temperature from 8 °C to room temperature.Thermal initiation polymerization and redox initiate polymerization of C/W HIPEs
For thermal initiation polymerization, stirring was further continued at 1000 rpm for 2 h after the formation of stable emulsions. The reaction vessel was heated to 70 °C rapidly. The temperature and pressure were maintained for 10 h and then the CO2 was slowly vented for more than 1 h.For redox initiation polymerization, when a stable emulsion was formed, stirring was further continued at 1000 rpm for 2 h before adding a controlled amount of tetramethylethylenediamine (TMEDA, 0.1 mL) at 150 bar. Stirring was ceased when TMEDA was mixed with emulsions thoroughly and the reaction vessel was then rapidly heated to 35 °C. The temperature and pressure were maintained for 2 h, and then CO2 was slowly vented for more than 1 h.
The shrinked product was recovered as a continuous, white monolithic sample that conformed to the internal dimensions of the reactor when immersed in water. Subsequently, the material was freeze-dried using a Scientz-12N freeze-dryer for more than 48 h.
Scanning electron microscope (SEM)
Porous PAM's morphology and porous structure was analyzed using an FEI Sirion 200 field emission scanning electron microscope (FE-SEM). For SEM analysis, the samples were mounted on aluminum stubs using adhesive graphite tape and sputter-coated with gold to make them conductive.In vitro cell culture
In vitro cell culture studies were conducted according to the literature.33,34 The ability of H9c2 cardiac muscle cells to grow into the PAM materials' 3D structure was assessed. In order to totally remove the residues, such as the monomer remaining inside the materials, we soaked the materials in deionized water for 2–3 days and changed the water every 8 h. The PAM materials were then cut into the suitable round shape and transferred into a 48-well plate and washed twice with ethanol to sterilise. The PAM materials were then washed at least twice with culture media to remove any residual ethanol and equilibrated in culture media (DMEM, 10% FBS, pen-strep) at 37 °C (12 h). For cell seeding, PAM cylinders were placed in 48 well plates. H9c2 cardiac muscle cell suspension (50 μL) containing 2.5 × 105 cells (unless stated otherwise) was added dropwise to each well containing PAM cylinder. The 48-well plates containing PAM materials and H9c2 cardiac muscle cells were then placed in an incubator for 2 h to equilibrate swelling and initial cell attachment. Afterwards, 1.0 mL of basal medium was added to each of the wells. An unseeded PAM was also placed in a well as a control sample. The cells were cultured in a CO2 incubator for 24 h, 48 h and 72 h at 37 °C, after which the PAM materials were fixed to assess cell proliferation using a fluorescence assay.Fluorescence assay
For further visualization of the growth and distribution of cells on the surface of each material, 4′,6-diamidino-2-phenylindole (DAPI) was used to stain cell nuclei. Samples were washed in PBS, stained with a solution of 10 μM of DAPI for 20 min, and rinsed in PBS prior to being imaged using an inverted fluorescence microscope (Nikon Eclipse Ti–S) in conjunction with NIS-Elements BR 3.1. Images were taken from the upper surface of the scaffold.To detect the cell adhesion and proliferation inside the material, wash the material three times (5 min each time) using PBS buffer before fixing with 10% formaldehyde for 90 min at room temperature, then the materials were transversely cut into slices using freezing microtome (Leica CM 1950) with the thickness ranging from 20 μm to 25 μm. Put slices on the glass slides and dry the slides completely before incubating the materials with 10 μM of DAPI by dropping on slices. Wash the slides carefully without disturbing the material slices and then nucleus of cell inside the materials could be observed by a Confocal microscopy (Olumpus81+FV1000).
The quantitative data about cell growth was obtained by counting cell numbers in 4–5 different regions, and the data are presented as mean of percentage ± SEM (Standard Error of Mean). The percentage of cell number refers to cell numbers counted in different time points including 36 h and 72 h normalized to 24 h, respectively.
Pore size analysis
Image J (1.46) was used to estimate the average pore size (d) and distribution by tracing a minimum of 300 pores for each sample.35 The pores were manually picked by built-in drawing functions and the area (A) of each pore was subsequently measured using the ‘measure’ function. The pore diameter was then calculated according to the following formula:
The pore size distribution of three typical samples were also recorded by mercury intrusion porosimetry using a MicroActive AutoPore V 9600 porosimeter. Samples were subjected to a pressure cycle starting at approximately 0.1 psi, increasing to 61000 psi in pre-defined steps to determine pore size, total intrusion volume, bulk density and porosity.
Results and discussion
Formation of stable C/W emulsions
Previous studies have shown that the presence of organic monomers, such as acrylamide, tend to have a destabilizing effect on C/W emulsions.20 This may be because of the adsorption of the monomers at the water–CO2 interface thus reducing interfacial tension and, therefore, stability of the emulsions.36 It may also be because of the fact monomers improve the solubility of the surfactant in the aqueous phase and are thus removed from the C/W interface.13 Therefore, the use of appropriate stabilizers to avoid above-mentioned issues are highly desired to form stable C/W emulsions. In our previous study,37 we synthesized various OVAc-based hydrocarbon surfactants, which have the ability to stabilize highly concentrated C/W emulsions. However, the industrial applications of such surfactants are limited due to their higher cost and fairly complex synthetic/purification protocols.In order to address these issues, we have used different concentration of PVA solution to investigate the emulsification of CO2 in an aqueous medium (40% w/v AM/MBAM in H2O, AM/MBAM = 8:2 w/w), as detailed in Table 1. Interestingly, these emulsions were found to be stable for a long period (at least 12 h) under static conditions (Fig. 1). These results suggested that the system is promising for emulsion templating approaches because the free-radical polymerization would be expected to occur before the emulsions become destabilized.
| PAM | Kind of PVA (hydrolyzed, g mol−1) | w/v (%) | Average pore diameterc (μm) |
|---|---|---|---|
| a Reaction conditions: AM + MBAM (40% w/v in H2O, AM/MBAM = 8:2 w/w), K2S2O8 (2% w/v based on monomer), PVA, 70 °C, 10 h, venting, and then freeze-drying.b Reaction conditions: AM + MBAM (40% w/v in H2O, AM/MBAM = 8:2 w/w), K2S2O8 (2% w/v based on monomer), PVA, TMEDA (0.1 mL at 150 bar), 35 °C, 2 h, venting, and then freeze-drying.c Average pore diameters were calculated using Image J software. |
|||
| 1a | 80%, 10000 |
1.25 | 26.67 ± 1.73 |
| 2a | 80%, 10000 |
2.50 | 22.53 ± 1.62 |
| 3a | 80%, 10000 |
3.75 | 21.12 ± 1.84 |
| 4a | 80%, 10000 |
5.00 | 18.14 ± 1.14 |
| 5a | 88%, 22000 |
2.50 | 15.67 ± 0.92 |
| 6a | 88%, 88000 |
2.50 | 21.21 ± 2.30 |
| 7b | 80%, 10000 |
1.25 | 13.18 ± 0.51 |
| 8b | 80%, 10000 |
2.50 | 13.36 ± 0.32 |
| 9b | 80%, 10000 |
3.75 | 15.18 ± 0.36 |
 | ||
| Fig. 1 Photographs of a PAM hydrogel formed by gelation of a C/W emulsion: (left) high-pressure reactor used in this experiment, (right) sample. | ||
PAM-based materials
In this study, partially hydrolyzed PVA stabilizers were used to stabilize C/W HIPEs at 8 °C. In the presence of PVA stabilizers, it was observed that the C/W HIPEs were sufficiently stable to act as templates for the formation of poly-HIPEs. In the thermally initiated polymerization, the reactor was quickly heated to 70 °C. The temperature and pressure were maintained for 10 h. Generally speaking, the stability of emulsions is decreased at higher temperature due their faster coalescence. The remarkable stability of the C/W emulsions, under the optimized conditions, was further demonstrated by polymerization of the continuous aqueous phase to produce porous, cross-linked PAM-based materials (Fig. 1). The resulting emulsion-templated materials were soaked in water to remove residual initiator and monomers, and then freeze-dried. When the freeze dried samples were placed in water, their shapes were recovered completely within a few seconds. The pore characterization data of the C/W emulsion-templated materials are summarized in Table 1.The structural features of the monoliths such as porosity, pore size and interconnectivity have a significant effect upon adhesion, growth and penetration of the cells within the 3D structures. In this study, SEM analysis was used to characterize the pore morphology of the emulsion-templated porous PAM materials. As shown in Fig. 2 and 3, the PAM materials possess high degree of porosity having an open porous structure. The cavities are interconnected by a series of channels, which enable the rapid transport of nutrients and oxygen through a polyHIPE material that is highly desired for cellular growth for tissue engineering applications.
 | ||
| Fig. 2 PAM-based porous materials synthesized by thermal-initiation polymerization with different contents of PVA (80%, 10000) stabilizers as characterized by scanning electron microscopy (left) and Image J software (right). The experimental details are listed in the Table 1. (a) Sample 1, 1.25%. (b) Sample 2, 2.50%. (c) Sample 3, 3.75%. (d) Sample 4, 5.00%. | ||
 | ||
| Fig. 3 PAM-based porous materials synthesized by thermal-initiation polymerization with different kinds of PVA stabilizers as characterized by scanning electron microscopy (left) and Image J software (right). The experimental details are listed in the Table 1. (a) Sample 5, PVA (88%, 22000) 2.50%. (b) Sample 6, PVA (88%, 88000) 2.50%. | ||
Effect of PVA stabilizer concentration
A series of C/W emulsion-templated porous materials were synthesized over a range of stabilizer concentration (1.25–5.00% w/v). All the variables, such as mixing time, mixing speed, monomer concentration, initiator concentration, CO2 volume, and CO2 pressure were kept constant. Fig. 2 shows a series of SEM images and pore diameter distribution plots for the emulsion-templated materials synthesized at increasing stabilizer concentration.Equivalent circle diameter of pores was calculated by using Image J software.35 The average pore size of PAM monoliths increased from 18.14 ± 1.14 μm to 26.67 ± 1.73 μm when the PVA concentration was decreased from 5.00% to 1.25% (Table 1, sample 1–4), which is in accordance with dominant micellar nucleation mechanism proposed by Harkins.38 According to the mechanism, an increase in the ratio of initial stabilizer to monomer results in an increase in the relative amount of micelles and thus the number of the CO2 droplets, hence decreasing the size of the CO2 droplets. After polymerization and venting of CO2, the size of cells decreases accordingly.
In order to complement the pore analysis by SEM, three typical samples (sample 1–3) were characterized by mercury intrusion porosimeter and the results are comparable. The data of average pore diameter, total intrusion volume, bulk density and porosity are provided in Table 2. The average size of the scaffolds pores is about 10–20 μm, and the whole porosity is around 90%, which shows the pores have good interconnectivity. The figures of the pore size distribution characterized by mercury intrusion porosimetry are provided in the Fig. 4.
| PAM | Average pore diameter (μm) | Total intrusion volume (mL g−1) | Bulk density (g mL−1) | Porosity (%) |
|---|---|---|---|---|
| 1 | 19.06 | 10.01 | 0.0903 | 90.37 |
| 2 | 16.79 | 9.62 | 0.0975 | 93.74 |
| 3 | 9.97 | 8.40 | 0.1065 | 89.46 |
 | ||
| Fig. 4 PAM-based porous materials synthesized by thermal-initiation polymerization with different contents of PVA (80%, 10000) stabilizers as characterized by mercury intrusion porosimetry. (a) Sample 1, 1.25%. (b) Sample 2, 2.50%. (c) Sample 3, 3.75%. | ||
In 2003, Butler also used perfluoropolyether (PFPE) ammonium carboxylate as surfactant to do the similar research.13 They added 1% w/v surfactant in this system by changing the PVA concentration. The average pore size of PAM monoliths increased from 4.3 μm to 9.5 μm when the PVA concentration was decreased from 5% to 1%. We also observed the similar trend i.e., a decrease in the pore size of materials by increasing the surfactant concentration.
The stabilizer concentration could influence the normalized pore size distribution. The results indicated that the pore size distribution would become broader with the decrease of stabilizer concentration (Fig. 2). The broad cell distribution could be because of the addition of small amount of stabilizer leading to the destabilization by bridging flocculation between the droplets.39 A single polymer molecule may adsorb onto two or more CO2 droplets, physically hold them together resulting in their coalescence to some extent. An attractive feature of this approach is that the pore size of the final materials is tunable simply by changing the stabilizer concentration.
Effect of different kinds of PVA
In our previous study,20 we have demonstrated that the solubility of the OVAc in CO2 is strongly dependent on the molecular weight because the OVAc molecular weight could affect the hydrophilic–CO2–philic balance of the C/W emulsions and thus affects the pore properties of the C/W emulsion-templated materials. But for partially hydrolyzed PVA, the affect is much more complicated. With an increase in the molecular weight of PVA, the viscosity of PVA solution in the same concentration is higher and the emulsions are more stable within a specific range of stabilizer concentration. There is thus an optimal increase in stability with increasing viscosity. As the viscosity begins to increase, the barrier to coalescence increases. Increasing the viscosity of the continuous phase also reduces the amount of dispersed phase that could be incorporated.40 However, by increasing the molecular weight of emulsifier, the solubility of the emulsifier in CO2 decreases dramatically. The compatibility between the PVA and CO2 decreases, and so does the emulsifier stability.It was observed that the more hydrolyzed PVA (88% hydrolyzed, 22000 g mol−1) showed better performance than the less hydrolyzed PVA (80% hydrolyzed, 10000 g mol−1). There are less VAc-groups in more hydrolyzed PVA (88% hydrolyzed, 22000 g mol−1) than the less hydrolyzed PVA (80% hydrolyzed, 10000 g mol−1) in the same concentration. The viscosity of the medium also plays a major role in the stabilization of emulsions. An increase in the viscosity of the dispersion medium increases the stability of the C/W emulsions leading to smaller and more kinetically stable CO2 emulsion droplets which are then translated into smaller cells and pores in the cross-linked PAM materials. Compared with the sample 2, the sample 5 has smaller pore size and relatively narrow pore size distribution (Fig. 3a). For sample 6, higher molecular weight of PVA (88% hydrolyzed, 88000 g mol−1) showed poor compatibility in CO2 leading to the larger pore size and relatively broader pore size distribution (Fig. 3b).
Effect of process of polymerization
In this study, we found that the emulsions prepared by PVA can be stable for a long time at 70 °C and a series of PAM-based emulsion-templated materials were synthesized.It is known that the solubility of CO2 decreases at higher temperature. The quicker the motion of its molecules with increasing temperature, the lesser would be the stability of C/W emulsions due their rapid coalescence but it does not usually proceed to the formation of two separate phases. So, in this report, we exploited the above fact by initiating polymerization of C/W emulsions in the presence of partially hydrolyzed PVA at lower temperature.
Butler et al. found that the materials produced by redox initiation were at least as porous (and often even more porous) than the equivalent samples produced using thermal initiation.13 In the redox initiated polymerization, a catalytic amount of a redox co-initiator (TMEDA) is used to initiate polymerization reactions at 35 °C. It was, therefore, possible to carry out the reactions using liquid CO2 at much lower reaction pressure. In general, in accordance with the Bulters' findings, the materials produced using redox initiation were at least as porous (and often more porous) than the equivalent samples produced using thermal initiation at 70 °C. The effect of the process of polymerization on the pore size distribution and median pore diameter is shown in Fig. 5. It is obvious that the pore size of these materials decreases, which suggests that the temperature plays an important role in the formation and pore tunability of these monoliths.
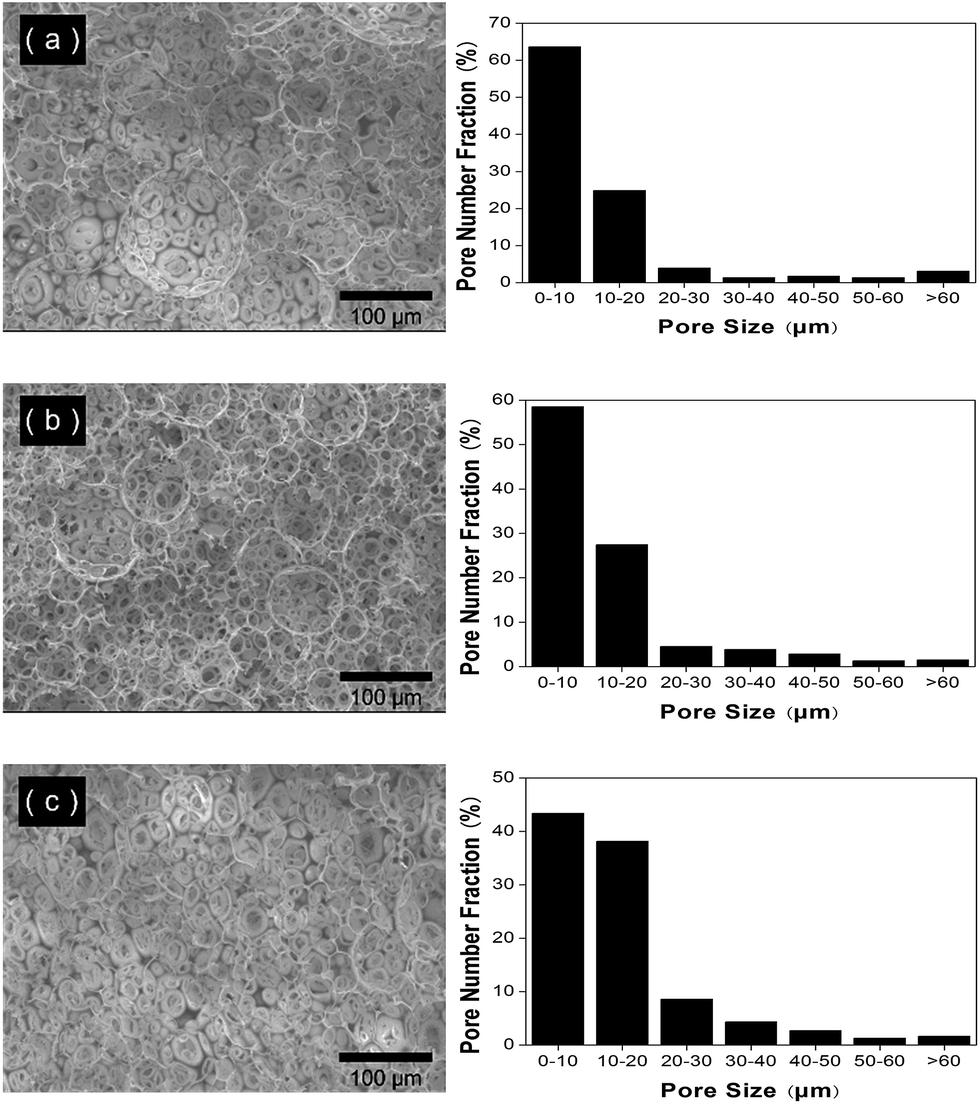 | ||
| Fig. 5 PAM-based porous materials synthesized by redox polymerization with different contents of PVA (80%, 10000) stabilizers as characterized by scanning electron microscopy (left) and Image J software (right). The experimental details are listed in the Table 1. (a) Sample 7 1.25%. (b) Sample 8, 2.50%. (c) Sample 9, 3.75%. | ||
In vitro cell proliferation using PAM materials
Porous, 3D scaffolds have been used extensively as biomaterials in the field of tissue engineering for in vitro study of cell–scaffold interactions and tissue synthesis and in vivo study of induced tissue and organ regeneration.41 Pore structure is an essential consideration in the development of scaffolds for tissue engineering. Pores must be interconnected to allow cell growth, migration and nutrient flow.42However, there are currently only a few successful applications of CO2/water emulsion polymerization template materials for cell culture due to the use of toxic and nonbiodegradable surfactants.43 In our previous study,21 we synthesized the amphiphilic cationic surfactant, PVAc-b-PDMAEMA, by successive RAFT/ATRP polymerization, and highly porous emulsion-templated PAM materials with tunable pore-size and open-pore size were subsequently prepared by the polymerization of the continuous phase of C/W emulsions. We also evaluated the potential of porous materials as scaffolds for promoting the growth of human embryonic lung diploid fibroblast cells, however, only a few cells were found on the surface indicating the limitation of such materials.
In this paper, we used commercially available partially hydrolyzed PVA to prepare C/W emulsions at low temperature to prepare porous emulsion-templated PAM materials. PVA is a water soluble polymer which is suitable for biological applications because of its biocompatibility.44–46 Sample 2 and sample 3 were chosen as scaffold to guide the cardiac myoblast cellular growth and the growth of cells on these materials can easily be observed (Fig. 6). After culturing for 72 h, the cell growth rate was increased to 470.7% and 481.6% for sample 2 and 3 respectively, compared to their growth at 24 h.
 | ||
| Fig. 6 Fluorescence imaging of the cell proliferation of culturing for (a) 24 h, (b) 48 h, (c) 72 h after cell seeding on sample 2 (top surfaces) and (d) 24 h, (e) 48 h, (f) 72 h after cell seeding on sample 3 (top surfaces). On the right side of the fluorescence figures are quantitation of H9c2 cells grown on samples from 24 h to 72 h. Data are presented as mean of percentage ± SEM. | ||
In order to get the further data regarding to the cell adhesion and proliferation in the 3D structure, sample 7 and sample 9 were chosen as a scaffold to guide the cardiac myoblast cellular growth. As shown in Fig. 7, H9c2 cells penetrated into the pores of PAM hydrogels. Through culturing for 72 hours, the percentage of cell number could reach 191% and 245% in sample 7 and 9 respectively normalized to 24 h.
 | ||
| Fig. 7 Fluorescence imaging of the cell proliferation of culturing for (a) 24 h, (b) 48 h, (c) 72 h after cell seeding on sample 7 (internal surfaces) and (d) 24 h, (e) 48 h, (f) 72 h after cell seeding on sample 9 (internal surfaces). On the right side of the fluorescence figures are quantitation of H9c2 cells grown on samples from 24 h to 72 h. Data are presented as mean of percentage ± SEM. | ||
The results suggested that PAM materials are suitable for cells growth due to their better cell adhesion and proliferation properties. These biocompatible porous materials with highly interconnected pore may be potential scaffolds for artificial organs. For example, the growth and proliferation of H9c2 cardiac muscle cells demonstrates the potential of such porous materials as scaffolds for artificial heart using 3D printing technology.
Conclusions
We have demonstrated the use of commercially available partially hydrolyzed PVA as a stabilizer for the synthesis of stable C/W emulsions. This process is much simpler, convenient and inexpensive compared to those involving the use of expensive and difficult-to-synthesize stabilizers, and would be useful for industrial-scale applications of C/W emulsions polymerization. Highly porous emulsion-templated materials with tunable cells size and interconnected pores were also prepared by the polymerization of the continuous phase of C/W emulsions. The influence of the stabilizer concentration and molecular weight of PVA and reaction conditions of the polymerization process on the morphology of the porous structures were also investigated. The SEM analysis confirms the interconnected pores in the size range of 13.18–26.67 μm. Moreover, we have also demonstrated the use of porous PAM materials as tissue engineering scaffolds. The H9c2 cardiac muscle cells can easily grow and proliferate on the surface of these porous PAM scaffolds demonstrating their to produce organs by 3D printing technology.Acknowledgements
The authors are grateful to the Analysis and Test Center of Huazhong University of Science & Technology for characterization of samples. This work is financially supported by the Program for New Century Excellent Talents in University (NCET-10-0389) and National Natural Science Foundation of China (21474033/51273074/51173058).Notes and references
- M. S. Silverstein, Polymer, 2014, 55, 304–320 CrossRef CAS.
- A. Barbetta and N. R. Cameron, Macromolecules, 2004, 37, 3202–3213 CrossRef CAS.
- S. Kovacic, N. B. Matsko, K. Jerabek, P. Krajnc and C. Slugovc, J. Mater. Chem. A, 2013, 1, 487–490 RSC.
- I. Pulko and P. Krajnc, Macromol. Rapid Commun., 2012, 33, 1731–1746 CrossRef CAS PubMed.
- X. Dong, Y. Wang, Y. Huang, J. Liu and X. Jing, J. Mater. Chem., 2011, 21, 16147–16152 RSC.
- H. K. He, W. W. Li, M. Lamson, M. J. Zhong, D. Konkolewicz, C. M. Hui, K. Yaccato, T. Rappold, G. Sugar, N. E. David, K. Damodaran, S. Natesakhawat, H. Nulwala and K. Matyjaszewski, Polymer, 2014, 55, 385–394 CrossRef CAS.
- F. Su, C. L. Bray, B. Tan and A. I. Cooper, Adv. Mater., 2008, 20, 2663–2666 CrossRef CAS PubMed.
- S. W. Zou, Z. J. Wei, Y. Hu, Y. H. Deng, Z. Tong and C. Y. Wang, Polym. Chem., 2014, 5, 4227–4234 RSC.
- W. Li, W. Zhang, X. Dong, L. Yan, R. Qi, W. Wang, Z. Xie and X. Jing, J. Mater. Chem., 2012, 22, 17445–17448 RSC.
- T. Zhang, Y. Wu, Z. Xu and Q. Guo, Chem. Commun., 2014, 50, 13821–13824 RSC.
- Y. P. Wu, T. Zhang, Z. G. Xu and Q. P. Guo, J. Mater. Chem. A, 2015, 3, 1906–1909 RSC.
- J. M. DeSimone, Science, 2002, 297, 799–803 CrossRef CAS PubMed.
- R. Butler, I. Hopkinson and A. I. Cooper, J. Am. Chem. Soc., 2003, 125, 14473–14481 CrossRef CAS PubMed.
- R. Butler, C. M. Davies and A. I. Cooper, Adv. Mater., 2001, 13, 1459–1463 CrossRef CAS.
- S. Partap, I. Rehman, J. R. Jones and J. A. Darr, Adv. Mater., 2006, 18, 501–504 CrossRef CAS.
- Z. Bing, J. Y. Lee, S. W. Choi and J. H. Kim, Eur. Polym. J., 2007, 43, 4814–4820 CrossRef CAS.
- C. Palocci, A. Barbetta, A. La Grotta and M. Dentini, Langmuir, 2007, 23, 8243–8251 CrossRef PubMed.
- B. Tan, J. Y. Lee and A. I. Cooper, Macromolecules, 2007, 40, 1945–1954 CrossRef CAS.
- J. Y. Lee, B. Tan and A. I. Cooper, Macromolecules, 2007, 40, 1955–1961 CrossRef CAS.
- K. Chen, N. Grant, L. Liang, H. Zhang and B. Tan, Macromolecules, 2010, 43, 9355–9364 CrossRef CAS.
- S. C. Zhang, W. Luo, W. Yan and B. Tan, Green Chem., 2014, 16, 4408–4416 RSC.
- E. J. Beckman, J. Supercrit. Fluids, 2004, 28, 121–191 CrossRef CAS.
- H. Lee, J. W. Pack, W. X. Wang, K. J. Thurecht and S. M. Howdle, Macromolecules, 2010, 43, 2276–2282 CrossRef CAS.
- N. A. Birkin, N. J. Arrowsmith, E. J. Park, A. P. Richez and S. M. Howdle, Polym. Chem., 2011, 2, 1293–1299 RSC.
- E. J. Park, A. P. Richez, N. A. Birkin, H. Lee, N. Arrowsmith, K. J. Thurecht and S. M. Howdle, Polymer, 2011, 52, 5403–5409 CrossRef CAS.
- S. C. Zhang, K. P. Chen, L. Y. Liang and B. Tan, Polymer, 2013, 54, 5303–5309 CrossRef CAS.
- S. C. Zhang, Y. L. Luo, H. W. Yang, H. J. Yang and B. Tan, Polym. Chem., 2013, 4, 3507–3513 RSC.
- S. S. Adkins, X. Chen, I. Chan, E. Torino, Q. P. Nguyen, A. W. Sanders and K. P. Johnston, Langmuir, 2010, 26, 5335–5348 CrossRef CAS PubMed.
- Z. Shen, M. A. McHugh, J. Xu, J. Belardi, S. Kilic, A. Mesiano, S. Bane, C. Karnikas, E. Beckman and R. Enick, Polymer, 2003, 44, 1491–1498 CrossRef CAS.
- D. Tapriyal, Y. Wang, R. M. Enick, J. K. Johnson, J. Crosthwaite, M. C. Thies, I. H. Paik and A. D. Hamilton, J. Supercrit. Fluids, 2008, 46, 252–257 CrossRef CAS.
- W. Luo, S. Zhang, P. Li, R. Xu, Y. Zhang, L. Liang, C. D. Wood, Q. Lu and B. Tan, Polymer, 2015, 61, 183–191 CrossRef CAS.
- X. Fan, V. K. Potluri, M. C. McLeod, Y. Wang, J. Liu, R. M. Enick, A. D. Hamilton, C. B. Roberts, J. K. Johnson and E. J. Beckman, J. Am. Chem. Soc., 2005, 127, 11754–11762 CrossRef CAS PubMed.
- N. Annabi, S. M. Mithieux, A. S. Weiss and F. Dehghani, Biomaterials, 2010, 31, 1655–1665 CrossRef CAS PubMed.
- M. Kollmer, V. Keskar, T. G. Hauk, J. M. Collins, B. Russell and R. A. Gemeinhart, Biomacromolecules, 2012, 13, 963–973 CrossRef CAS PubMed.
- X. Zhong and F. Dehghani, Green Chem., 2012, 14, 2523–2533 RSC.
- C. T. Lee, P. A. Psathas, K. P. Johnston, J. deGrazia and T. W. Randolph, Langmuir, 1999, 15, 6781–6791 CrossRef CAS.
- B. Tan and A. I. Cooper, J. Am. Chem. Soc., 2005, 127, 8938–8939 CrossRef CAS PubMed.
- W. D. Harkins, J. Am. Chem. Soc., 1947, 69, 1428–1444 CrossRef CAS PubMed.
- E. Dickinson, M. Golding and M. J. W. Povey, J. Colloid Interface Sci., 1997, 185, 515–529 CrossRef CAS PubMed.
- H. H. Chen and E. Ruckenstein, J. Colloid Interface Sci., 1991, 145, 260–269 CrossRef CAS.
- F. J. O'Brien, B. A. Harley, I. V. Yannas and L. J. Gibson, Biomaterials, 2005, 26, 433–441 CrossRef PubMed.
- I. V. Yannas, Clin. Mater., 1992, 9, 179–187 CrossRef CAS PubMed.
- N. Annabi, J. W. Nichol, X. Zhong, C. Ji, S. Koshy, A. Khademhosseini and F. Dehghani, Tissue Eng., 2010, 16, 371–383 CrossRef CAS PubMed.
- G. Fussell, J. Thomas, J. Scanlon, A. Lowman and M. Marcolongo, J. Biomater. Sci., Polym. Ed., 2005, 16, 489–503 CrossRef CAS PubMed.
- Z. S. Nurkeeva, G. A. Mun, A. V. Dubolazov and V. V. Khutoryanskiy, Macromol. Biosci., 2005, 5, 424–432 CrossRef CAS PubMed.
- B. E. Jensen, A. A. Smith, B. Fejerskov, A. Postma, P. Senn, E. Reimhult, M. Pla-Roca, L. Isa, D. S. Sutherland, B. Stadler and A. N. Zelikin, Langmuir, 2011, 27, 10216–10223 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |