
Influence of carbon nanofillers on the curing kinetics of epoxy-amine resin
Abstract
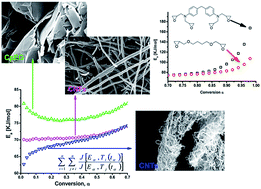
The cure kinetics of an epoxy resin based on the tetrafunctional epoxy precursor N,N′-tetraglycidyl methylene dianiline-(TGMDA) hardened with 4,4-diaminodiphenyl sulfone is investigated. The influence of carbon nanofillers (carbon nanotubes, carbon nanofibers, and graphene based nanoparticles) on the cure kinetic is studied. Kinetic analysis is performed by dynamic and isothermal differential scanning calorimetry (DSC). In dynamic experiments, the activation energy was computed using an advanced isoconversional method while under isothermal conditions, the Kamal’s model of diffusion control was applied to simulate the systems throughout the curing process. The isothermal analysis shows that the introduction of the diluent decreases, particularly the activation energy of secondary amine-epoxy reaction. A similar effect was obtained by the dynamic DSC analysis that shows a decrease in the activation energy for α > 0.7, a value of conversion for which it is considered that the reaction of secondary amines is active. The inclusion in the resin of one-dimensional fillers does not lead to big differences in the curing kinetics behaviour with respect to the raw epoxy. An increase in the activation energy is found in the case of highly exfoliated graphite. This is likely due to a reduction of free molecular segments of the epoxy network trapped inside the self-assembly structures.
 Please wait while we load your content...
Please wait while we load your content...

