
Electrochemical degradation of perfluorooctanoic acid (PFOA) by Yb-doped Ti/SnO2–Sb/PbO2 anodes and determination of the optimal conditions†
Abstract
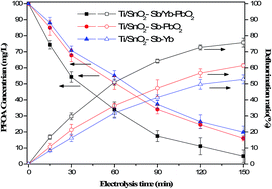
Model aqueous solutions of perfluorooctanoic acid (PFOA, 100 mg L−1) were electro-oxidized in a homemade container. The electrocatalytic behavior and anodic performance of Ti/SnO2–Sb/Yb–PbO2, Ti/SnO2–Sb–PbO2 and Ti/SnO2–Sb–Yb anodes in sodium electrolytes were compared. The SnO2–Sb/Yb–PbO2 anode demonstrated better electrocatalytic performance compared with the SnO2–Sb–PbO2 and SnO2–Sb–Yb electrodes in terms of both degradation and defluorination. Then a systematic experimental study was designed as follows to analyze the influencing factors: initial concentration of PFOA (10 mg L−1 to 200 mg L−1), current density (1 mA cm−2 to 40 mA cm−2), initial pH value (3 to 11) and electrode distance (5 mm to 20 mm). After a 150 min electrolysis, the optimum reaction conditions were obtained and the degradation and defluorination ratios reached 95.11 ± 3.9% and 75.7 ± 2.8%, respectively. Under the optimum conditions, the degradation of PFOA followed pseudo-first-order kinetics (0.0193 min−1) and the degradation half-life was 35.9 min. The produced F− was measured using a fluoride ion selective electrode, whereas the intermediate PFCAs with short-chain lengths were measured using HPLC-MS. A detailed degradation pathway was proposed in this study by analyzing the intermediates and the recovery of fluoride.
 Please wait while we load your content...
Please wait while we load your content...

