
Exploring the metallic phase of N2O under high pressure
Chunye Zhua,
Haixin Bia,
Shoutao Zhanga,
Shubo Weia and
Quan Li*ab
aState Key Laboratory of Superhard Materials, Jilin University, Changchun 130012, China. E-mail: liquan777@jlu.edu.cn; Tel: +86-431-85167557
bCollege of Materials Science and Engineering, Jilin University, Changchun 130012, China
First published on 27th July 2015
Abstract
Simple molecular solids are expected to undergo structural phase transitions to form frameworks or polymeric solids under high pressure. The high-pressure structures of N2O have long attracted considerable attention. We combine the CALYPSO searching method with density functional theory to investigate the phase stabilities and structural changes of N2O at high pressure. We find two metallic structures of N2O which may be observed in high-pressure experiments. The current calculations reveal that the C2/m is the most stable structure over a pressure range of 177–194 GPa, and the other C2/c is metastable and only 10 meV per atom higher in energy than the C2/m structure at 180 GPa. At higher pressure, the metallic C2/m phase transforms into an insulating phase with space group of P21/m.
1. Introduction
Pressure is a thermodynamic parameter of paramount importance for chemical equilibria and chemical kinetics. Under high pressure, the interatomic distances become shorter and the bonding patterns of materials can be altered, causing profound effects on numerous physical and chemical properties.1 High pressure plays an important role in the synthesis of new materials. Synthetic diamond is one of classic examples of high-pressure application.2 Moreover, the pressure can also effectively lower the barrier of chemical reaction and can be used to synthesize compounds with distinct species, such as NaCl3.3Simple molecular solids are characterized by strong covalent intramolecular interaction and weak van der Waals intermolecular interaction. At high pressure, the distance between atoms becomes shorter, and the intermolecular interactions of molecular crystals become highly repulsive which increases the instability of electrons localized within intramolecular bonds. This leads to unexpected transformations which molecular solids get through a full reorganization of the chemical bond connectivity to reduce free energy such as ionization,4–8 polymerization,9–11 metallization12,13 and the like. The new classes of materials may exhibit entirely new electronic, optical, and physical properties.14,15 For example, pressure-induced polymerization of molecular crystals with super hardness,16 superconductivity,17 and high energy density9,18 have been found.
N2O is one of the most extensively studied molecular crystals for its applications in medicine, refrigeration, and combustion reaction. The physical properties of N2O and CO2 are very similar, such as isoelectronic, linear molecules, and the nearly melting point. Although N2O has no inversion symmetric since the O atom is located at one end, it has been shown that N2O molecules solidify into a random head-to-tail orientation disorder at low pressures.19,20 N2O is therefore expected to form similar structures with CO2 at low pressures. In the solid state, the N2O molecules crystallize into the same configurations as CO2,21 such as Pa![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) (ref. 22) structure at ambient condition and Cmca structure above ∼5 GPa.19,20,23 Early experiments have found the solid N2O decomposes into an ionic crystal NO+NO3− and compressed N2 molecules at high pressure and temperature.6,7 Recently, theoretical study of N2O has predicted that N2O forms a one-dimensional polymer with an all-nitrogen backbone analogous to cis-polyacetylene in which alternate N atoms are bonded to O atoms above 60 GPa.24 Later on, the studied pressure range is up to 500 GPa,25 and a new N2O nanotube structure is found to be the most stable form above 180 GPa. Generally, molecular solids will go through insulator–metal transition at sufficiently high pressures due to the broadening of electronic bands,15,26 e.g., solid O2 transforms to metallic phase near 95 GPa13 and iodine undergoes a pressure-induced insulator–metal transition near 16 GPa.27 However, the metallic N2O have not been found so far. With the evolution of computer simulation technology, useful theoretical research can be as an aid in the interpretation of experimental data and guide experiment.28 Here, we perform our swarm structural searching method29–31 combined with first-principles calculations to explore structures and physical properties of N2O under high pressure.
(ref. 22) structure at ambient condition and Cmca structure above ∼5 GPa.19,20,23 Early experiments have found the solid N2O decomposes into an ionic crystal NO+NO3− and compressed N2 molecules at high pressure and temperature.6,7 Recently, theoretical study of N2O has predicted that N2O forms a one-dimensional polymer with an all-nitrogen backbone analogous to cis-polyacetylene in which alternate N atoms are bonded to O atoms above 60 GPa.24 Later on, the studied pressure range is up to 500 GPa,25 and a new N2O nanotube structure is found to be the most stable form above 180 GPa. Generally, molecular solids will go through insulator–metal transition at sufficiently high pressures due to the broadening of electronic bands,15,26 e.g., solid O2 transforms to metallic phase near 95 GPa13 and iodine undergoes a pressure-induced insulator–metal transition near 16 GPa.27 However, the metallic N2O have not been found so far. With the evolution of computer simulation technology, useful theoretical research can be as an aid in the interpretation of experimental data and guide experiment.28 Here, we perform our swarm structural searching method29–31 combined with first-principles calculations to explore structures and physical properties of N2O under high pressure.
2. Calculation methods
The structure searching was performed based on the particle swarm optimization algorithm as implemented in CALYPSO (crystal structure analysis by Particle Swarm Optimization).29–31 The method unbiased by any known structural information has been demonstrated by recent successes in predicting structures of various systems.32–35 The underlying ab initio structural relaxations and the electronic band structure calculations were performed within the framework of density functional theory (DFT) as implemented by the VASP (Vienna Ab initio Simulation Package) code.36 The calculations were carried out at the generalized gradient approximation (GGA)37 level using the Perdew–Burke–Ernzerh (PBE) of exchange correlation functional. The electronic wave functions were expanded in a plane-wave basis set with a cutoff energy of 1000 eV for all cases. The electron–ion interaction was described by means of projector augmented wave (PAW)38 pseudopotential with 2s22p3 and 2s22p4 electrons as valence for N and O atoms, respectively. The Heyd–Scuseria–Ernzerhof (HSE)39 hybrid functional is used to achieve an accurate electronic band dispersion of stable phases. We employed the Bader charge analysis approach40 for evaluating the charge transfer. Monkhorst-Pack k-point41 meshes with a grid of 0.03 Å−1 for Brillouin zone sampling were chosen to achieve the total energy convergence of less than 1 meV per atom. The phonon dispersion curves were computed by the direct supercell calculation method as implemented in the Phonopy program.423. Results and discussion
We have performed structure prediction simulations for N2O with variable-cell simulation cell sizes of 1–4 and 6 formula units (f.u.) at 40–300 GPa. The enthalpy–pressure relations of various interesting structures are shown in Fig. 1. Analysis of our simulation results has confirmed the experimental Cmca structure and the earlier predicted 2D polymer (Pnma),25 cis-polymer (Pnma),24 and nanotube structure (P21/m).25 In addition, two exotic metallic structures: C2/c (8 f.u. per cell) and C2/m (4 f.u. per cell) were uncovered here. The Cmca structure transforms to the cis-polymer structure at about 57 GPa, in good agreement with previous calculations.24 However, our results clarify and correct previous structural assignments at high pressures: the C2/m structure of N2O becomes energetically preferable to cis-polymer structure above 177 GPa. Above 194 GPa, N2O forms a nanotube P21/m structure. We found that a C2/c structure is favored over the cis-polymer structure above 186 GPa. It should be note that C2/c structure is metastable phase and the enthalpy of C2/c structure is only 10 meV per atom higher in energy than that of C2/m structure at 180 GPa. Below we focus on these two new phases which may be synthesized at high pressure.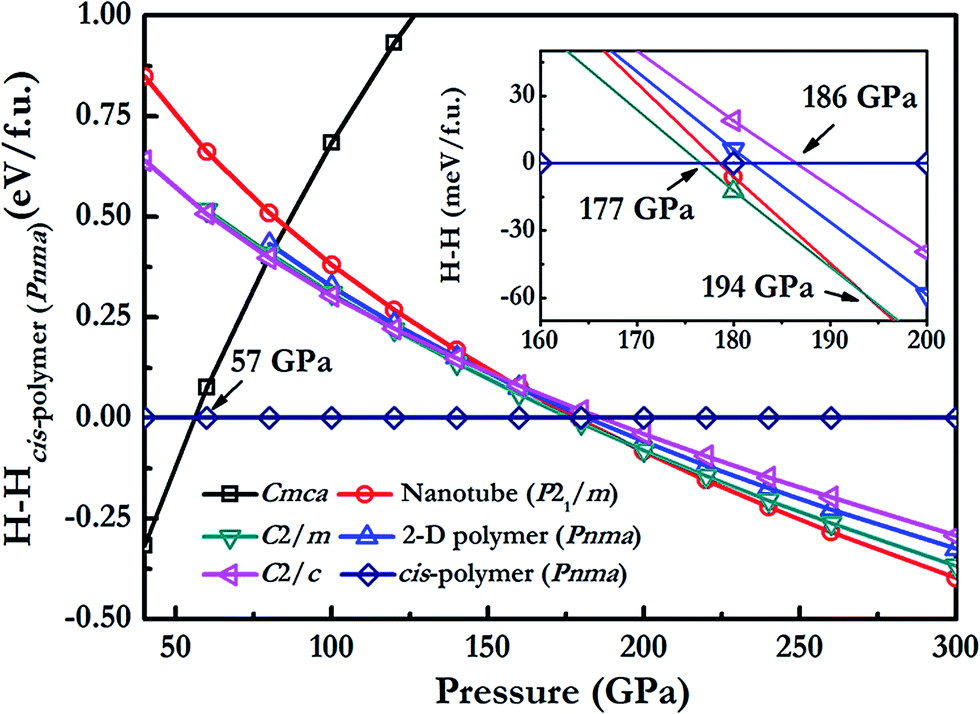 | ||
| Fig. 1 The enthalpies per formula unit of various structures as a function of pressure with respect to cis-polymer structure of N2O. | ||
The crystal structures of C2/c and C2/m are shown in Fig.2, which are both layered structures. Layered structures also appear in the polymerized solids of other molecules, such as N2,43 CO,44 and CO2.45 There are two types of N atom present in both C2/c and C2/m structure: one is N1 connected with both N atom and O atom, and the other is N2 only connected with N atom. At 180 GPa, from Fig. 2a, the layer of C2/c can be viewed as bulbs stacking. Every N atom is bonded with three neighbouring atoms, while each O atom is bonded with one N atom. The unit cell of the C2/c structure has 8 f.u. per cell with parameters of a = 4.954 Å, b = 3.668 Å, c = 7.819 Å and β = 125.333°, with O atoms at Wyckoff 8f position (−0.2084, −0.0016, 0.0766) and N atoms occupying two inequivalent 8f positions: (0.3384, −0.0036, 0.2172) and (0.3082, −0.1692, 0.8443). The C2/m structure possesses similar bonding states with that of the C2/c structure. The N atoms in the same layer form wrinkled N6 rings. (Fig. 2c.). It is interesting that the C2/m structure of N2O is related to the theoretically predicted Cmcm structure of solid CO,44 which also contains six-membered C6 rings. At 180 GPa, the optimized lattice parameters of C2/m are a = 8.556 Å, b = 2.183 Å, c = 3.188 Å and β = 75.539° with O atoms at Wyckoff 4i position (0.1787, 0.5, 0.7631) and N atoms occupying two inequivalent 4i positions: (0.0258, 0, 0.2908) and (0.8755, 0.5, 0.8459). The interlayer distance decreases significantly with pressure while the intralayer structure is hardly affected.
 | ||
| Fig. 2 The C2/c (up) and C2/m (down) structures at 200 GPa. (a) Top view of C2/c structure along the layered stacking direction. (b) Top view of C2/c structure along b-axis. (c) Top view of C2/m structure along the layered stacking direction. (d) Top view of C2/m structure along b-axis. | ||
To investigate the dynamical stabilities of our predicted N2O structures, the phonon dispersions of C2/c and C2/m structures are shown in Fig. 3. No imaginary phonon frequencies are found in these two structures, indicating the dynamical stability. The cells of C2/c and C2/m contain 24 and 12 atoms, giving 72 and 36 phonon branches, respectively. The phonon bands of C2/m have very little dispersion along Y–A and E–C directions (which is the interlayer directions), showing that the corresponding interactions are weak.
 | ||
| Fig. 3 Phonon dispersion relations of (a) C2/c at 200 GPa, (b) C2/m at 180 GPa. | ||
The electronic band structure of C2/c and C2/m at 180 GPa are shown in Fig. 4. Due to the PBE calculations severely underestimate the band gaps, we also include band calculations using the HSE hybrid functional for comparison. Both PBE and HSE calculations reveal the metallic nature of C2/c and C2/m phases. For the C2/m phases, the dispersions of the electron bands are strong in the intralayers (e.g. along the Γ–Y direction), but very weak between the layers along the Y–A and E–C directions. This observation indicates that in-layer interactions are much stronger than the inter-layer direction.
 | ||
| Fig. 4 Electronic band structures at 180 GPa for (a) C2/c and (b) C2/m. | ||
Chemical bonding behavior is the key to get a full understanding of the metallic nature of these structures. The nature of their bonding was probed by calculating the electron localization functions (ELF)46 of Fig. 5. The ELF is a measure of relative electron localization, and large ELF values indicate there is a high tendency of electron pairing, such as cores, bonds, and lone pairs. The high electron localization can be seen in the region between adjacent N–N and N–O bonds in C2/c and C2/m, indicative of covalent bonding. Fig. 6 shows the bond lengths of C2/c and C2/m as a function of pressure. For the C2/m structure, the bond lengths of N1–N2 are remarkably larger than N2–N2 bond lengths. At 180 GPa, the bond length between two N2 atoms is 1.292 Å, prominently shorter than the N1–N2 bond length of 1.383 Å. By comparison with the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond in the HN3 molecule (1.23 Å),47 the N2–N2 bond can be reasonably classified as the double bond and the N1–N2 bond as the single bond. We employed the Bader charge analysis approach for evaluating the actual charge transfer between N and O atoms. The calculated O Bader charges are 6.45e. The Bader charges indicate a substantial charge transfer from N to O, illustrating the mixture of ionic and covalent bonds appear between the N and O atoms. Each O atom forms a single N–O bond and accepts one electron from N1 atom to satisfy the octet rule. N1 atom loses one electron and forms three single bonds, and N2 atom forms two single bonds and one double bond. In consequence, every N has one remaining electron forming a set of π bonds spanning all N atoms in the same layer. The delocalized π electrons can be free to move throughout the same layer, and give rise to property of conductivity, which is similar to graphite. For the C2/c structure, the bond lengths of N1–N2 are nearly close to N2–N2 bond lengths. From Fig. 6a, the N1–N2 bond length dramatically decreases with the increasing pressure. At 260 GPa, N1–N2 bond length become shorter than the N2–N2 bond lengths. That is because the direction of N2–N2 bond is nearly parallel to the layers, while the direction of N1–N2 bond is along the layers stacking direction (Fig. 2b). The weak interaction of the inter-layer direction leads to the easier compression of N1–N2 bond. For C2/c structure at 180 GPa, the N–O bonding behavior is similar to that in C2/m structure. The calculated N2–N2 bond length is 1.309 Å and there are two types of N1–N2 bond with alternate distances of 1.321 Å and 1.323 Å. The valence bond description suggests alternating single and double bonds. This bonding pattern also forms the delocalized π electrons which is responsible of metallic nature of C2/c structure. At 194 GPa, the C2/m structure transforms into the P21/m structure.25 The P21/m structure is proposed by the earlier theoretical prediction. In this structure, O atom bonded with two N atoms, and every N atom has a lone pair with two single N–N bonds and one single N–O bond. The covalent bonds and lone-pair electrons are together the driving force for the insulating character of P21/m structure, since the electrons are highly localized. The metal–insulator transition is previously exemplified in dense lithium,48 sodium,49 and oxygen.50,51 In contrast to oxygen, the metal–insulator transition pressure (1.9 TPa) is considerably higher than 194 GPa in the current N2O.
N double bond in the HN3 molecule (1.23 Å),47 the N2–N2 bond can be reasonably classified as the double bond and the N1–N2 bond as the single bond. We employed the Bader charge analysis approach for evaluating the actual charge transfer between N and O atoms. The calculated O Bader charges are 6.45e. The Bader charges indicate a substantial charge transfer from N to O, illustrating the mixture of ionic and covalent bonds appear between the N and O atoms. Each O atom forms a single N–O bond and accepts one electron from N1 atom to satisfy the octet rule. N1 atom loses one electron and forms three single bonds, and N2 atom forms two single bonds and one double bond. In consequence, every N has one remaining electron forming a set of π bonds spanning all N atoms in the same layer. The delocalized π electrons can be free to move throughout the same layer, and give rise to property of conductivity, which is similar to graphite. For the C2/c structure, the bond lengths of N1–N2 are nearly close to N2–N2 bond lengths. From Fig. 6a, the N1–N2 bond length dramatically decreases with the increasing pressure. At 260 GPa, N1–N2 bond length become shorter than the N2–N2 bond lengths. That is because the direction of N2–N2 bond is nearly parallel to the layers, while the direction of N1–N2 bond is along the layers stacking direction (Fig. 2b). The weak interaction of the inter-layer direction leads to the easier compression of N1–N2 bond. For C2/c structure at 180 GPa, the N–O bonding behavior is similar to that in C2/m structure. The calculated N2–N2 bond length is 1.309 Å and there are two types of N1–N2 bond with alternate distances of 1.321 Å and 1.323 Å. The valence bond description suggests alternating single and double bonds. This bonding pattern also forms the delocalized π electrons which is responsible of metallic nature of C2/c structure. At 194 GPa, the C2/m structure transforms into the P21/m structure.25 The P21/m structure is proposed by the earlier theoretical prediction. In this structure, O atom bonded with two N atoms, and every N atom has a lone pair with two single N–N bonds and one single N–O bond. The covalent bonds and lone-pair electrons are together the driving force for the insulating character of P21/m structure, since the electrons are highly localized. The metal–insulator transition is previously exemplified in dense lithium,48 sodium,49 and oxygen.50,51 In contrast to oxygen, the metal–insulator transition pressure (1.9 TPa) is considerably higher than 194 GPa in the current N2O.
 | ||
| Fig. 5 Calculated electron localization functions (ELF) for (a) C2/c at 180 GPa, and (b) C2/m at 180 GPa. | ||
 | ||
| Fig. 6 Calculated bond lengths as a function of pressure. (a) C2/c and (b) C2/m. | ||
4. Conclusions
Using the CALYPSO method for crystal structure prediction combined with first-principle calculations, two novel metallic structures of N2O with space group C2/m and C2/c were discovered. The C2/m structure becomes the most stable phase above 177 GPa and it transforms into an insulate P21/m structure above 194 GPa. Two metallic structures are both layered structures, and every N atom is threefold-coordinated, while each O atom is only bonded with one N atom. The metallic behavior of N2O is derived from delocalized π electrons. The N2O structures can be tuned with increasing pressure from insulating behavior (Pnma phases), to melallic (C2/m) and then back to insulating (P21/m). The predicted formation of metallic structure in N2O represents a significant step forward in understanding the behavior of N2O and other molecular crystals at high pressures.Acknowledgements
This work was supported by the China 973 Program (2011CB808200), the Natural Science Foundation of China under No. 51202084, 11474125, and 11274136, the 2012 Changjiang Scholars Program of China, Changjiang Scholarand Innovative Research Team in University (IRT1132). Parts of the calculations were performed in the High Performance Computing Center (HPCC) of Jilin University.References
- W. Grochala, R. Hoffmann, J. Feng and N. W. Ashcroft, Angew. Chem., Int. Ed., 2007, 46, 3620 CrossRef CAS PubMed.
- F. Bund, J. Chem. Phys., 1963, 38, 631 CrossRef PubMed.
- W. Zhang, A. R. Oganov, A. F. Goncharov, Q. Zhu, S. E. Boulfelfel, A. O. Lyakhov, E. Stavrou, M. Somayazulu, V. B. Prakapenka and Z. Konopkova, Science, 2013, 342, 1502 CrossRef CAS PubMed.
- Y. Wang, H. Liu, J. Lv, L. Zhu, H. Wang and Y. Ma, Nat. Commun., 2011, 2, 563 CrossRef PubMed.
- C. J. Pickard and R. Needs, Nat. Mater., 2008, 7, 775 CrossRef CAS PubMed.
- M. Somayazulu, A. Madduri, A. F. Goncharov, O. Tschauner, P. F. McMillan, H.-k. Mao and R. J. Hemley, Phys. Rev. Lett., 2001, 87, 135504 CrossRef CAS.
- C. S. Yoo, V. Iota, H. Cynn, M. Nicol, J. H. Park, T. Le Bihan and M. Mezouar, J. Phys. Chem. B, 2003, 107, 5922 CrossRef CAS.
- A. Y. Kuznetsov, L. Dubrovinsky, A. Kurnosov, M. M. Lucchese, W. Crichton and C. A. Achetel, Adv. Phys. Chem., 2009, 2009, 180784 Search PubMed.
- M. J. Lipp, W. J. Evans, B. J. Baer and C.-S. Yoo, Nat. Mater., 2005, 4, 211–215 CrossRef CAS PubMed.
- M. I. Eremets, A. G. Gavriliuk, I. A. Trojan, D. A. Dzivenko and R. Boehler, Nat. Mater., 2004, 3, 558 CrossRef CAS PubMed.
- V. Iota, C. Yoo and H. Cynn, Science, 1999, 283, 1510 CrossRef CAS.
- Y. Fujii, K. Hase, N. Hamaya, Y. Ohishi, A. Onodera, O. Shimomura and K. Takemura, Phys. Rev. Lett., 1987, 58, 796 CrossRef CAS.
- Y. Akahama, H. Kawamura, D. Häusermann, M. Hanfland and O. Shimomura, Phys. Rev. Lett., 1995, 74, 4690 CrossRef CAS.
- V. Schettino and R. Bini, Phys. Chem. Chem. Phys., 2003, 5, 1951 RSC.
- R. J. Hemley, Annu. Rev. Phys. Chem., 2000, 51, 763 CrossRef CAS PubMed.
- C. Yoo, H. Cynn, F. Gygi, G. Galli, V. Iota, M. Nicol, S. Carlson, D. Häusermann and C. Mailhiot, Phys. Rev. Lett., 1999, 83, 5527 CrossRef CAS.
- N. Ashcroft, Phys. Rev. Lett., 2004, 92, 187002 CrossRef CAS.
- C. Mailhiot, L. Yang and A. McMahan, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 14419 CrossRef CAS.
- R. Mills, B. Olinger, D. Cromer and R. Lesar, J. Chem. Phys., 1991, 95, 5392 CrossRef CAS PubMed.
- B. Kuchta, R. Etters and R. LeSar, J. Chem. Phys., 1992, 97, 5662 CrossRef CAS PubMed.
- K. Aoki, H. Yamawaki, M. Sakashita, Y. Gotoh and K. Takemura, Science, 1994, 263, 356 CAS.
- W. C. Hamilton and M. Petrie, J. Phys. Chem., 1961, 65, 1453 CrossRef CAS.
- V. Iota, J. Park and C. Yoo, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 064106 CrossRef.
- H. Xiao, Q. An, W. A. Goddard, W.-G. Liu and S. V. Zybin, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 321 Search PubMed.
- Q. An, H. Xiao, W. A. Goddard III and X. Meng, J. Phys. Chem. Lett., 2014, 5, 485 CrossRef CAS.
- C.-S. Yoo, Phys. Chem. Chem. Phys., 2013, 15, 7949 RSC.
- A. Balchan and H. Drickamer, J. Chem. Phys., 1961, 34, 1948 CrossRef CAS PubMed.
- E. Zurek and W. Grochala, Phys. Chem. Chem. Phys., 2015, 17, 2917 RSC.
- Y. Wang, J. Lv, L. Zhu and Y. Ma, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 094116 CrossRef.
- Y. Wang, J. Lv, L. Zhu and Y. Ma, Comput. Phys. Commun., 2012, 183, 2063 CrossRef CAS PubMed.
- Y. Wang, M. Miao, J. Lv, L. Zhu, K. Yin, H. Liu and Y. Ma, J. Chem. Phys., 2012, 137, 224108 CrossRef PubMed.
- L. Zhu, H. Liu, C. J. Pickard, G. Zou and Y. Ma, Nat. Chem., 2014, 6, 644 CAS.
- S. Lu, Y. Wang, H. Liu, M.-s. Miao and Y. Ma, Nat. Commun., 2014, 5, 3666 CAS.
- Q. Li, D. Zhou, W. Zheng, Y. Ma and C. Chen, Phys. Rev. Lett., 2013, 110, 136403 CrossRef.
- M. Zhang, H. Liu, Q. Li, B. Gao, Y. Wang, H. Li, C. Chen and Y. Ma, Phys. Rev. Lett., 2015, 114, 015502 CrossRef.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207 CrossRef CAS PubMed.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188 CrossRef.
- A. Togo, F. Oba and I. Tanaka, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 134106 CrossRef.
- J. Sun, D. D. Klug, R. Martoňák, J. A. Montoya, M.-S. Lee, S. Scandolo and E. Tosatti, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 6077 CrossRef CAS PubMed.
- J. Sun, D. D. Klug, C. J. Pickard and R. J. Needs, Phys. Rev. Lett., 2011, 106, 145502 CrossRef.
- Y. Ma, A. R. Oganov, Z. Li, Y. Xie and J. Kotakoski, Phys. Rev. Lett., 2009, 102, 065501 CrossRef.
- A. D. Becke and K. E. Edgecombe, J. Chem. Phys., 1990, 92, 5397 CrossRef CAS PubMed.
- J. Evers, M. Gobel, B. Krumm, F. Martin, S. Medvedyev, G. Oehlinger, F. X. Steemann, I. Troyan, T. M. Klapotke and M. I. Eremets, J. Am. Chem. Soc., 2011, 133, 12100 CrossRef CAS PubMed.
- J. Neaton and N. Ashcroft, Nature, 1999, 400, 141 CrossRef CAS.
- Y. Ma, M. Eremets, A. R. Oganov, Y. Xie, I. Trojan, S. Medvedev, A. O. Lyakhov, M. Valle and V. Prakapenka, Nature, 2009, 458, 182 CrossRef CAS PubMed.
- L. Zhu, Z. Wang, Y. Wang, G. Zou, H.-k. Mao and Y. Ma, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 751 CrossRef CAS PubMed.
- J. Sun, M. Martinez-Canales, D. D. Klug, C. J. Pickard and R. J. Needs, Phys. Rev. Lett., 2012, 108, 045503 CrossRef.
| This journal is © The Royal Society of Chemistry 2015 |