
Direct synthesis of β-ketophosphonates and vinyl phosphonates from alkenes or alkynes catalyzed by CuNPs/ZnO†
Victoria Gutierrez‡
a,
Evangelina Mascaró‡a,
Francisco Alonsob,
Yanina Moglie*a and
Gabriel Radivoy*a
aInstituto de Química del Sur, INQUISUR (CONICET-UNS), Departamento de Química, Universidad Nacional del Sur, Av. Alem 1253, 8000 Bahía Blanca, Argentina. E-mail: gradivoy@criba.edu.ar
bInstituto de Síntesis Orgánica (ISO) and Departamento de Química Orgánica, Facultad de Ciencias, Universidad de Alicante, Apdo. 99, E-03080, Alicante, Spain
First published on 28th July 2015
Abstract
Copper nanoparticles (CuNPs) supported on ZnO have been shown to effectively catalyze the direct synthesis of β-ketophosphonates from alkenes or alkynes, and that of vinyl phosphonates from alkynes and diethylphosphite, under air and in the absence of any additive or ligand. When using alkynes as starting materials, the selectivity proved to be dependent on the nature of the alkyne. Thus, alkynes conjugated with an aromatic ring or a carbon–carbon double bond gave β-ketophosphonates as the main reaction products, whereas aliphatic alkynes or alkynes conjugated with a carbonyl group led to the formation of the corresponding vinyl phosphonates.
Introduction
Phosphorus containing organic compounds are of widespread interest in synthetic organic chemistry. This is due to the remarkable biological activities of many phosphorus derivatives which make them of potential interest and application in diverse fields, ranging from medicinal chemistry to the agrochemical industry. Phosphonic acids and esters are widely known as extremely important structural analogues to the corresponding carboxylic acids and esters.1 Among this family of organophosphorus compounds, β-ketophosphonates have received considerable attention in recent years, not only due to their varied biological activities2 and metal-complexing abilities,3 but also because they are valuable precursors for the construction of α,β-unsaturated compounds (Horner–Wadsworth–Emmons reaction)4 and for the synthesis of chiral β-amino-5 and β-hydroxy6 phosphonic acids.Several methods for the synthesis of β-ketophosphonates can be found in the scientific literature.7 The most common approaches are based in the condensation between carboxylic acid derivatives and alkyl phosphonates,8 and in the hydration of alkynyl phosphonates,9 both protocols having some important drawbacks; while condensation reactions often need for the use of strong bases and/or cryogenic conditions, many of the hydration-based methods require the use of toxic mercury salts in combination with acids or bases. Although recently published procedures based on the use of Pd10 or Au11 catalysts for the hydration of alkynylphosphonates circumvent the use of toxic reagents, there is still a need for the development of direct synthetic methodologies to access β-ketophosphonates from simple and readily available starting materials. In this regard, the transition metal-catalysed oxyphosphorylation of alkynes with H-phosphonates has very recently been tackled by two independent research groups. In one of these works, Zhao et al.12 reported a one-pot strategy for the synthesis of β-ketophosphonates using AgNO3/CuSO4·5H2O as catalyst and K2S2O8 as oxidant additive. The reactions are conducted under air and in CH2Cl2–H2O as solvent. In a more recent paper, Song et al.,13 have communicated the oxyphosphorylation of alkynes or alkynyl carboxylic acids with H-phosphonates, catalysed by CuOTf/FeCl3 in DMSO, under O2 atmosphere, in the presence of Et3N as base.
On the other hand the copper/iron co-catalyzed oxyphosphorylation of alkenes with dioxygen and H-phosphonates published by Wei and Ji,14 is, to the best of our knowledge, the only transition metal-catalyzed direct synthesis of β-ketophosphonates from alkenes that has been published so far.
Even though the above mentioned transition metal-catalysed methodologies are useful and attractive, in most cases long reaction times (24 h), oxygen atmosphere and/or the use of different additives is required.
Vinyl phosphonates represent another prominent member of the family of phosphorous derivatives, they are well known as important synthetic intermediates15 and have diverse applications as monomers and co-monomers in polymeric materials.16 Most of the existing methods for P–C(vinylic) bond formation are based on palladium-catalyzed reactions,1,17 which usually require the use of phosphine ligands. Alternatively, the use of Cu(I)18 or Ni(II)19 catalysts have also been reported but both methodologies need for harsh reaction conditions or phosphine ligands to give the desired vinyl phosphonates in reasonable yields.
In the last years we have actively been working in the development of new and mild methodologies based on the use of bare or supported copper nanoparticles (CuNPs) for their application in the construction of C–C and C–heteroatom bonds.20 During the course of our investigations about CuNPs-catalysed C–P coupling reactions, we have found that the reaction between aryl acetylenes or terminal alkenes with commercial diethyl phosphite, catalysed by CuNPs supported on ZnO, led to the direct formation of β-ketophosphonates. Notably, when aliphatic alkynes were reacted under the same reaction conditions, the corresponding vinyl phosphonates were obtained instead as the major products. We want to present herein our results on the development of a new and simple methodology that allow the direct synthesis of β-ketophosphonates and vinyl phosphonates from alkenes or alkynes as simple and readily available starting materials, under air atmosphere and mild reaction conditions, catalysed by CuNPs/ZnO.
Results and discussion
We started our study by choosing phenylacetylene (1a) as model substrate to optimize the reaction conditions. The reactions were performed under air atmosphere and in the absence of any other additive or ligand. Initially, the reaction of 1a with (EtO)2P(O)H was tested in the presence of CuNPs supported on a variety of organic and inorganic materials using acetonitrile as the solvent (Table 1). All the CuNPs based catalysts were prepared according to the methodology previously reported by us,20 the CuNPs (3–6 nm in size) being synthesized by fast reduction of anhydrous CuCl2 with an excess of lithium sand and a catalytic amount of DTBB (4,4′-di-tert-butylbiphenyl) as electron carrier. Fig. 1 shows a TEM image and size distribution graphic for CuNPs/ZnO catalyst (see ESI† for experimental details and characterization of the catalyst). As can be seen, well dispersed spherical copper nanoparticles with an average particle size of 6.0 ± 0.5 nm are obtained.
|
||
|---|---|---|
| Entry | Catalyst | Conversionb (%) |
| a Reaction conditions: phenylacetylene (1 mmol), diethyl phosphite (2 mmol), catalyst (40 mg, 1.7 mol% Cu), in acetonitrile (2 mL) at 70 °C under air atmosphere, 6 h.b Determined by GC using an internal standard.c Reaction performed using 60 mg of catalyst.d Reaction performed using 20 mg of catalyst.e Conversion obtained at 24 h of reaction time. | ||
| 1 | CuNPs/ZnO | 86, 83c, 69d |
| 2 | CuNPs/CeO2 | 52 |
| 3 | CuNPs/PVP | 42 |
| 4 | CuNPs/cellulose | 28 |
| 5 | CuNPs/Celite® | 14 |
| 6 | CuNPs/MWNT | 40 |
| 7 | CuNPs/MCM-41 | 38 |
| 8 | CuCl2/ZnO | 83e |
| 9 | CuCl/ZnO | 39e |

 | ||
| Fig. 1 TEM image and size distribution graphic of CuNPs in CuNPs/ZnO catalyst. | ||
As shown in Table 1, the best results were obtained by working at 70 °C and using ZnO as support for the CuNPs (Table 1, entry 1). The lower conversions obtained when using any of the other supports listed in Table 1, could suggest the necessity for the presence of Lewis acid (Zn2+) and/or Lewis basic (O2−) sites on the support for the reaction to take place at a reasonable rate.
The optimal amount of catalyst was found to be 40 mg (see Table 1, footnotes c and d), giving a copper loading of 1.7 mol% referred to the starting alkyne. Interestingly, we found that when using CuCl2 as copper source, a similar conversion to the β-ketophosphonate product was obtained, although in a significantly longer reaction time (Table 1, entry 8). On the contrary, the use of CuCl/ZnO as catalyst demonstrated to be less effective (Table 1, entry 9).
Control experiments carried out, both in the absence of the CuNPs/ZnO catalyst or in the presence of the support or the CuNPs alone, gave no conversion of the starting phenylacetylene to the desired product.
Then, the effect of the solvent, added bases and reaction atmosphere was studied. Among the different solvents tested acetonitrile demonstrated to be the solvent of choice. Other solvents such as methanol, dimethyl sulfoxide, dichloromethane, toluene, acetone and tetrahydrofuran gave almost no conversion of the starting material, whereas a 15% conversion to the alkyne homocoupling product was observed when water was used as the reaction medium. The reaction carried out under N2 atmosphere gave low conversion to the desired product 2a (near 15%). The addition of a base (Cs2CO3, Na2CO3, TMDA or triethylamine) proved counterproductive for the reaction, giving no conversion of the starting alkyne after 24 h of reaction time.
Another interesting observation was that the suspension of the catalyst in the solvent was kept for those cases in which no conversion was measured. On the contrary, for those reactions which progressed to the desired products, the catalyst was completely dissolved in the reaction media, forming a single clear phase.
Under the optimized conditions, a series of aromatic and aliphatic terminal alkynes were then tested. As can be seen in Table 2, terminal aryl acetylenes gave the corresponding β-ketophosphonates 2a–h as the major reaction products in good to excellent yields. Other dialkyl phosphites such as dimethyl- or dibutyl phosphite, were also suitable phosphorylating agents (Table 2, compounds 2a′ and 2a′′). Interestingly, 1-ethynylcyclohex-1-ene gave the corresponding β-ketophosphonate in good yield (Table 2, compound 2h), thus suggesting that the presence of a conjugated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C in the starting alkyne is mandatory for the formation of the β-ketophosphonate products.
C in the starting alkyne is mandatory for the formation of the β-ketophosphonate products.


When aliphatic alkynes were subjected to the same reaction conditions, the anti-Markovnikov vinyl phosphonates 2i–m (Table 2) were formed as the main reaction products, together with minor amounts of the corresponding β-ketophosphonates. In all cases, an inseparable mixture of E/Z isomers was obtained. It must be pointed out that alkyne 1m, which is not aliphatic but conjugated with a CO, gave the corresponding vinylphosphonate 2m as the main reaction product, whereas, as above mentioned, alkyne 1h which is conjugated with a CC bond gave the corresponding β-ketophosphonate product (2h). This selectivity dependent on the alkyne nature has not been observed in previously reported copper-catalysed oxyphosphorylation of alkynes, and represents a very important feature of this CuNPs/ZnO catalyst. On the other hand, when internal alkynes were tested, both diaryl- and dialkyl alkynes, as well as aryl alkyl ones, low conversions to a complex mixture of products were obtained.
Then, we decided to test the reaction of alkenes with diethyl phosphite under the same optimized reaction conditions. We observed that only terminal alkenes were converted to the corresponding β-ketophosphonates (Table 3). It is worthy of note that the CuNPs/ZnO catalyst demonstrated not to be compatible with the presence of hydroxyl or carboxylic acid groups in the starting alkyne or alkene. These functional groups could form strong hydrogen-bonds with the Lewis basic sites at the catalyst support, thus preventing its participation in the reaction pathway. All of the above experimental observations would suggest that the ZnO support could be playing a non-innocent role in the catalytic system, probably through the P–H bond activation.21
|
|---|
| a Isolated yield after chromatographic purification (hexane/AcOEt). |
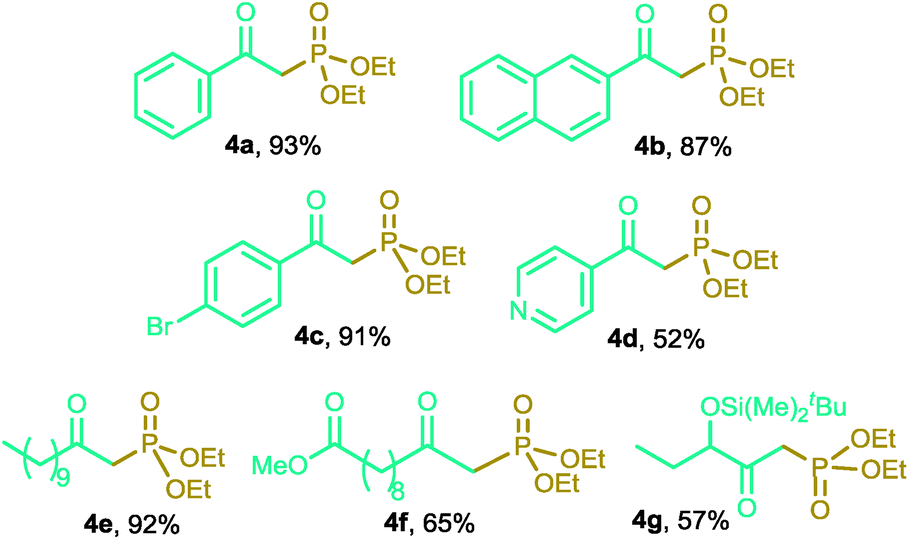 |

We conducted a series of additional experiments in order to get some information about the reaction mechanism involved in each of the transformations studied. Initially, based on our experimental observations and previous reports, we studied the effect of the addition of TEMPO as radical scavenger. Under the optimized conditions, the presence of TEMPO inhibited the formation of β-ketophosphonates, both starting from phenylacetylene (1a) or styrene (3a), suggesting a radical pathway for the mechanism of these reactions. On the contrary, the transformation of 1-octyne into the corresponding vinyl phosphonate 2i, was not affected by the addition of TEMPO to the reaction mixture. On the other hand, the reaction of 1-octyne under the optimized conditions but carried out under N2 atmosphere, gave the vinyl phosphonate product 2i with almost the same conversion and in a similar reaction time to that of the same reaction conducted under air. As above mentioned, the synthesis of β-ketophosphonate 2a from phenylacetylene failed when it was carried out in the absence of air. The use of an O2 atmosphere, both for the reaction of 1-octyne and phenylacetylene, proved counterproductive.22
With the aim to get information on the oxidation state of copper in the catalyst, XPS analyses were performed, unfortunately the XPS spectra obtained were not conclusive, mainly due to a low signal-to-noise ratio that could be attributed to the low concentration of copper in the catalyst. Despite this, based on our previous works20 and the results showed in Table 1 (entries 8 and 9), we assume that copper(II) is most likely to be the active species in the catalyst.
Although the exact mechanism involved in the transformations studied is not clear enough at this stage, based on our experimental observations and previous reports by other authors,12,13 we suppose that a radical oxyphosphorylation process is more likely to occur in the synthesis of β-ketophosphonates. As shown in Fig. 2, Cu(II) species in combination with ZnO would be promoting the formation of the phosphorus radical III (Fig. 2). Addition of this radical intermediate to the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C or CC bond, and subsequent reaction with an active-oxygen copper complex, formed by reaction of CuNPs with dioxygen present in air, would led to reactive hydroperoxide intermediates.12,13 Finally, these reactive intermediates would evolve to the corresponding β-ketophosphonate products, through a keto–enol taumerism in the case of alkynes, and by elimination of water in the case of alkenes.
C or CC bond, and subsequent reaction with an active-oxygen copper complex, formed by reaction of CuNPs with dioxygen present in air, would led to reactive hydroperoxide intermediates.12,13 Finally, these reactive intermediates would evolve to the corresponding β-ketophosphonate products, through a keto–enol taumerism in the case of alkynes, and by elimination of water in the case of alkenes.
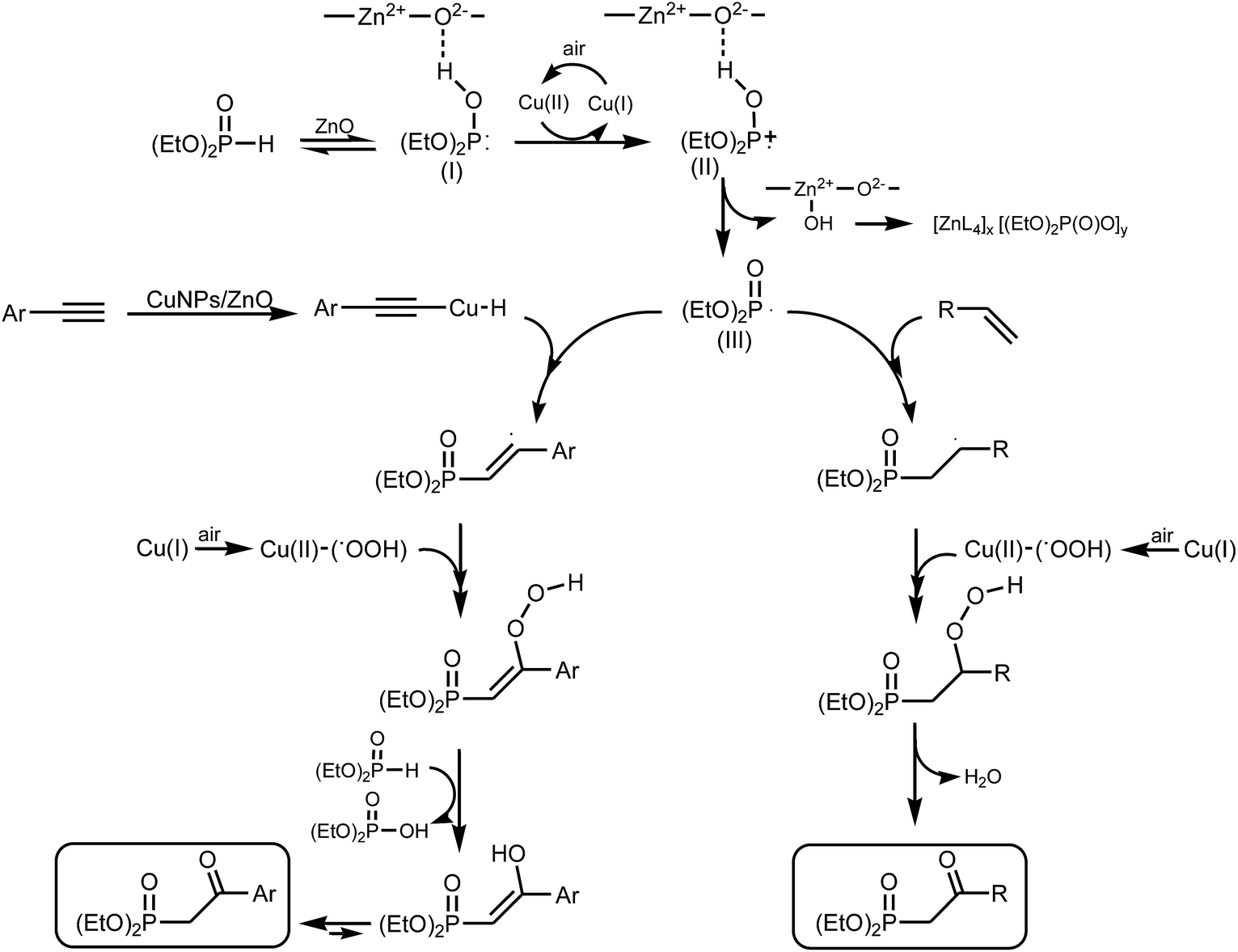 | ||
| Fig. 2 Proposed mechanistic pathway for the synthesis of β-ketophosphonates from aromatic alkynes and alkenes catalysed by CuNPs/ZnO. | ||
On the other hand, the observed formation of vinyl phosphonates from aliphatic alkynes, which was not inhibited by the presence of TEMPO (and also works in the absence of air), should be occurring through a non-radical process, probably due to the lower stability of the corresponding vinyl radical intermediate. Instead, a copper-catalysed anti-Markovnikov hydrophosphorylation of the aliphatic alkyne leading to the corresponding vinyl phosphonates, could take place. It is noteworthy that most of the reported methods for the metal-catalysed hydrophosphorylation of alkynes are based on the use of expensive and/or toxic palladium, nickel or rhodium catalysts,17c–e whereas copper-based ones have remained almost unexplored to date.23 With the aim to confirm this hypothesis, we carried out the reaction by using 1-deuterio-oct-1-yne as starting alkyne. As expected, the corresponding deuterated-vinyl phosphonate product was obtained, thus showing that, in the case of aliphatic alkynes, copper acetylides would not be formed under the reaction conditions and the reaction is more likely to occur via a copper-catalysed hydrophosphorylation process.
Conclusions
In conclusion, we have developed a new mild, simple and economical methodology for the direct synthesis of β-ketophosphonates and vinyl phosphonates from commercial or readily available starting materials, catalysed by CuNPs/ZnO. The catalyst showed to be very efficient working under air atmosphere and in the absence of any additive or ligand. Alternative copper sources such as CuCl2 in combination with ZnO, can also be utilized, however longer reaction times are needed. Although the exact mechanistic pathway is difficult to ascertain at this stage, based on our experimental observations, we propose that a copper-catalysed radical oxyphosphorylation mechanism would be involved in the formation of β-ketophosphonates, whereas a non-radical copper-catalysed hydrophosphorylation process would be more likely to occur in the formation of vinyl phosphonates. Furthermore, it should not be ruled out a bifunctional mode of action by the CuNPs/ZnO catalyst, where the CuNPs could be activating the C–H and/or the CC bond of the starting alkyne and the ZnO activating the diethyl phosphite P–H bond. Further mechanistic details and other useful synthetic applications of this catalytic system are now under study. The simplicity and high atom economy of this new methodology will surely be of wide interest among synthetic organic chemists.
Acknowledgements
This work was generously supported by the Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT) and Universidad Nacional del Sur (UNS) from Argentina. V. G. thanks the CONICET for a postdoctoral fellowship.Notes and references
- C. S. Demmer, N. Krogsgaard-Larsen and L. Bunch, Chem. Rev., 2011, 111, 7981 CrossRef CAS PubMed.
- (a) C. Balg, S. P. Blais, S. Bernier, J. L. Huot, M. Couture, J. Lapointe and R. Chênevert, Bioorg. Med. Chem., 2007, 15, 295 CrossRef CAS PubMed; (b) S. K. Perumal, S. A. Adediran and R. F. Pratt, Bioorg. Med. Chem., 2008, 16, 6987 CrossRef CAS PubMed; (c) M. D. Erion, H. Jiang and S. H. Boyer, US Patent, 20060046980, 2006.
- (a) G. Sturtz, J. C. Clement, A. Daniel, J. Molinier and M. Lenzi, Bull. Soc. Chim. Fr., 1981, 167 Search PubMed; (b) F. A. Cotton and R. A. Schunn, J. Am. Chem. Soc., 1963, 85, 2394 CrossRef CAS; (c) D. J. McCabe, E. N. Duesler and R. T. Paine, Inorg. Chem., 1985, 24, 4626 CrossRef CAS.
- (a) S. E. Kelly, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon, Oxford, 1991, vol. 1, pp. 729–818 and references cited therein Search PubMed; (b) W. S. Wadsworth Jr and W. D. Emmons, J. Am. Chem. Soc., 1961, 83, 1733 CrossRef; (c) J. Boutagy and R. Thomas, Chem. Rev., 1974, 74, 87 CrossRef CAS; (d) B. E. Maryanoff and A. B. Reitz, Chem. Rev., 1989, 89, 863 CrossRef CAS.
- (a) A. Ryglowski and P. Kafarski, Tetrahedron, 1996, 52, 10685 CrossRef CAS; (b) F. Palacios, C. Alonso and J. M. de los Santos, Chem. Rev., 2005, 105, 899 CrossRef CAS PubMed; (c) R. Kadyrov, J. Holz, B. Schäffner, O. Zayas, J. Almena and A. Börner, Tetrahedron: Asymmetry, 2008, 19, 1189 CrossRef CAS PubMed.
- (a) M. Kitamura, M. Tokunaga and R. Noyori, J. Am. Chem. Soc., 1995, 117, 2931 CrossRef CAS; (b) M. Chavez, M. Vargas, A. Suarez, E. Alvarez and A. Pizzano, Adv. Synth. Catal., 2011, 353, 2775 CrossRef CAS PubMed; (c) X. Tao, W. Li, X. Ma, X. Li, W. Fan, L. Zhu, X. Xie and Z. Zhang, J. Org. Chem., 2012, 77, 8401 CrossRef CAS PubMed.
- (a) B. A. Arbuzov, Pure Appl. Chem., 1964, 9, 307 CAS; (b) A. K. Bhattacharka and G. Thyagarajan, Chem. Rev., 1981, 81, 415 CrossRef; (c) F. Orsini, E. Di Teodoro and M. Ferrari, Synthesis, 2002, 1683 CrossRef CAS; (d) F. Orsini and A. Caselli, Tetrahedron Lett., 2002, 43, 7259 CrossRef CAS; (e) M. Koprowski, D. Szymanska, A. Bodzioch, B. Marciniak, E. Rozycka-Sokołowska and P. Bałczewski, Tetrahedron, 2009, 65, 4017 CrossRef CAS PubMed.
- (a) K. Narkunan and M. Nagarajan, J. Org. Chem., 1994, 59, 6386 CrossRef CAS; (b) R. R. Milburn, K. McRae, J. Chan, J. Tedrow, R. Larsen and M. Faul, Tetrahedron Lett., 2009, 50, 870 CrossRef CAS PubMed; (c) K. M. Maloney and J. Y. L. Chung, J. Org. Chem., 2009, 74, 7574 CrossRef CAS PubMed; (d) F. Palacios, A. M. Ochoa de Retanam and J. M. Alonso, J. Org. Chem., 2006, 71, 6141 CrossRef CAS PubMed; (e) J. Westermann, M. Schneider, J. Platzek and O. Petrov, Org. Process Res. Dev., 2007, 11, 200 CrossRef CAS.
- (a) B. Iorga, F. Eymery, D. Carmichael and P. Savignac, Eur. J. Org. Chem., 2000, 3103 CrossRef CAS; (b) A. J. Poss and R. K. Belter, J. Org. Chem., 1987, 52, 4810 CrossRef CAS; (c) G. Peiffer and P. Courbis, Can. J. Chem., 1974, 52, 2894 CrossRef CAS; (d) E. J. Corey and S. C. Virgil, J. Am. Chem. Soc., 1990, 112, 6429 CrossRef CAS; (e) S. D. Guile, J. E. Saxton and M. Thornton-Pett, J. Chem. Soc., Perkin Trans. 1, 1992, 1763 RSC.
- X. Li, G. Hu, P. Luo, G. Tang, Y. Gao, P. Xu and Y. Zhao, Adv. Synth. Catal., 2012, 354, 2427 CrossRef CAS PubMed.
- L. Xie, R. Yuan, R. Wang, Z. Peng, J. Xiang and W. He, Eur. J. Org. Chem., 2014, 2671 Search PubMed.
- X. Chen, X. Li, X.-L. Chen, L.-B. Qu, J.-Y. Chen, K. Sun, Z.-D. Liu, W.-Z. Bi, Y.-Y. Xia, H.-T. Wu and Y.-F. Zhao, Chem. Commun., 2015, 51, 3846 RSC.
- M. Zhou, M. Chen, Y. Zhou, K. Yang, J. Su, J. Du and Q. Song, Org. Lett., 2015, 17, 1786 CrossRef CAS PubMed.
- W. Wei and J.-X. Ji, Angew. Chem., Int. Ed., 2011, 50, 9097 CrossRef CAS PubMed.
- (a) X. Y. Jiao and W. G. Bentrude, J. Org. Chem., 2003, 68, 3303 CrossRef CAS PubMed; (b) M. Maffei, Curr. Org. Synth., 2004, 1, 355 CrossRef CAS; (c) M. N. Noshi, A. El-awa, E. Torres and P. L. Fuchs, J. Am. Chem. Soc., 2007, 129, 11242 CrossRef CAS PubMed. For review articles, see: (d) T. Minami and J. Motoyoshiya, Synthesis, 1992, 333 CrossRef CAS; (e) P. Adler, A. Fadel and N. Rabasso, Tetrahedron, 2014, 70, 4437 CrossRef CAS PubMed.
- (a) G. J. Schlichting, J. L. Horan, J. D. Jessop, S. D. Nelson, S. Seifert, Y. Yang and A. M. Herring, Macromolecules, 2012, 45, 3874 CrossRef CAS; (b) J. Xu, K. Jung, A. Atme, S. Shanmugam and C. Boyer, J. Am. Chem. Soc., 2014, 136, 5508 CrossRef CAS PubMed; (c) Q. Wu and R. A. Weiss, Polymer, 2007, 48, 7558 CrossRef CAS PubMed.
- (a) M. Kalek, A. Ziadi and J. Stawinski, Org. Lett., 2008, 10, 4637 CrossRef CAS PubMed; (b) N. S. Gulykina, T. M. Dolgina, G. N. Bondarenko and I. P. Beletskaya, Russ. J. Org. Chem., 2003, 39, 847 CrossRef. For review articles about the hydrophosphorylation of alkynes see: (c) F. Alonso, I. P. Beletskaya and M. Yus, Chem. Rev., 2004, 104, 3079 CrossRef CAS PubMed; (d) L. Coudray and J. L. Montchamp, Eur. J. Org. Chem., 2008, 3601 CrossRef CAS PubMed; (e) V. P. Ananikov, L. L. Khemchyan and I. P. Beletskaya, Synlett, 2009, 2375 CrossRef CAS.
- (a) D. Gelman, L. Jiang and S. L. Buchwald, Org. Lett., 2003, 5, 2315 CrossRef CAS PubMed; (b) S. Thielges, P. Bisseret and J. Eustache, Org. Lett., 2005, 7, 681 CrossRef CAS PubMed.
- (a) G. Trostyanskaya, D. Y. Titskiy, E. A. Anufrieva, A. A. Borisenkko, M. A. Kazankova and I. P. Beletskaya, Russ. Chem. Bull., Int. Ed., 2001, 50, 2095 CrossRef; (b) L. B. Han, C. Zhang, H. Yazawa and S. Shimada, J. Am. Chem. Soc., 2004, 126, 5080 CrossRef CAS PubMed.
- For some recent works, see: (a) F. Nador, M. A. Volpe, F. Alonso, A. Feldhoff, A. Kirschning and G. Radivoy, Appl. Catal., A, 2013, 455, 39 CrossRef CAS PubMed; (b) F. Alonso, Y. Moglie, G. Radivoy and M. Yus, J. Org. Chem., 2013, 78, 5031 CrossRef CAS PubMed; (c) F. Nador, M. A. Volpe, F. Alonso and G. Radivoy, Tetrahedron, 2014, 70, 6082 CrossRef CAS PubMed.
- M. Hosseini-Sarvari and M. Tavakolian, New J. Chem., 2012, 36, 1014 RSC.
- As suggested by one referee, the reaction under O2 atmosphere was tested. When oct-1-yne was used as starting alkyne, after 8 h of reaction time, a very low conversion to octanoic acid as the only organic product was observed, together with a large amount of diethylphosphate, produced by oxidation of diethylphosphite. On the other hand, when the reaction was carried out using phenylacetylene as starting alkyne, a 30% conversion to the corresponding β-ketophosphonate was observed, together with benzoic acid and other phosphorylated by-products whose structures could not be elucidated.
- I. G. Trostyanskaya and I. P. Beletskaya, Tetrahedron, 2014, 70, 2556 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, characterization data, and copies of 1H, 13C and 31P NMR spectra of all phosphonate products. Full characterization of catalyst is also included. See DOI: 10.1039/c5ra10223e |
| ‡ These authors contributed equally to this manuscript. |
| This journal is © The Royal Society of Chemistry 2015 |