
Simultaneous electrochemical determination of acetaminophen, caffeine and ascorbic acid using a new electrochemical sensor based on CuO–graphene nanocomposite†
Z. Monsef Khoshhesab*
Department of Chemistry, Payame Noor University, 19395-4697, Tehran, I.R. of Iran. E-mail: monsef_kh@pnu.ac.ir
First published on 30th October 2015
Abstract
A new nanocomposite based on CuO nanoparticles/graphene nanosheets was prepared and used as a new electrode material for the simultaneous determination of acetaminophen, caffeine and ascorbic acid. CuO nanoparticles were supported on graphene nanosheets by a simple method. This nanostructure was characterized by different techniques including scanning electron microscopy, X-ray diffraction, energy dispersive X-ray spectroscopy and Fourier transform infrared spectroscopy. The high electrochemical activity, fast electron transfer rate, high surface area and good antifouling properties of the synthesized nanostructure enhanced the oxidation peak currents and reduced the peak potentials of acetaminophen, caffeine and ascorbic acid at the surface of the proposed sensor. Simultaneous determination of analytes was explored using differential pulse voltammetry. A linear range of 0.025–5.3 μmol L−1 was achieved for acetaminophen, caffeine and ascorbic acid with detection limits of 0.008, 0.010 and 0.011 μmol L−1, respectively. Finally, the proposed method was used for their determination in blood serum, urine and pharmaceutical samples.
Introduction
Acetaminophen (AC), known as paracetamol, is commonly used as an antipyretic and analgesic medicine, and is considered safe at therapeutic levels for humans with normal drug use.1 Overdoses of this drug lead to the accumulation of toxic metabolites, which may cause hepatotoxicity and nephrotoxicity.1,2 Because AC is being increasingly used for therapeutic purposes, its determination and quality control are of vital importance.Caffeine (CA) is an active alkaloid component present in coca nuts, coca-cola, coffee and tea leaves.3 It is known to have many pharmacological effects including gastric acid secretion, diuretic, cardiac stimulant and stimulant of central nervous system.3,4 The stimulant effect of CA usually results in an increased ability for mental activity and muscular work.4 When taken in a reasonable amount, it reduces a desire for sweets by simulating the production of adrenal hormones 5 that cause blood sugar to be increased. The weakness, depression and discomfort from excess of alcohol can be cancelled out with black coffee or hypodermic injections of CA. Further, it helps in preventing a positive energy balance and obesity. It is also an accepted drug for intramuscular applications to treat arterial hypotension.3 Drugs consisting of AC and CA combination are mostly used as pain relief, central nervous system stimulant and an analgesic agent. An overdose of these combination drugs leads to vomiting, irregular heartbeat, nausea, cardiovascular diseases and cancer.3,6,7 Thus, their determination and quantification are much important in analgesic formulations and also can give beneficial guidance to human health and life.
Ascorbic acid (vitamin C, AA) is regarded as the most important water-soluble antioxidant in human plasma and mammalian cells which have mechanisms to recycle and accumulate it against a concentration gradient, suggesting that the vitamin might also have important intracellular functions. In biological systems, AA is a potent reducing agent and scavenger of free radicals.8–10 Most animals are able to synthesis vitamin C from glucose, but humans and other primates, lack the last enzyme involved in the synthesis of vitamin C (gulonolactone oxidase) and so require the presence of the vitamin in their diet. For human, lack of vitamin C in the diet can even cause death. Clinical data show that when ascorbate is given orally, fasting plasma concentrations are tightly controlled at <100 μmol L−1.8–10 For this purpose, AA is widely used as antioxidant agent in foods, drinks, and pharmaceutical products.
The large scale therapeutic uses of AC, CA and AA require fast, simple and sensitive methods to be developed for their determination in human body fluids, food samples and pharmaceutical preparations. Many analytical techniques have been described the determination of AC, CA and AA, including spectrophotometry, high-performance liquid chromatography, and electrochemical methods,11–13 among which, electrochemical method based modified electrodes have attracted more attention for their high sensitivity, simplicity, reproducibility, on site monitoring and low cost.
Nowadays, due to the need for the improvements in sensing characteristics of electrodes, such as selectivity, stability, and cost-effectiveness, new sensing layers has been studied to improve detection in chemical sensing and biosensing. Therefore, the improvements in components of sensing layer, for example, through incorporation of nanomaterials, can greatly enhance the analytical performance of chemical sensors and biosensors.14–18 With this strategy, the analytical performance of sensors has improved in sensitivity, selectivity, limit of detection (LOD), and signal to-noise ratio.14–20 In electrochemical applications, carbon based nanomaterials have been widely used for preparation of modified electrodes. Carbon nanomaterials such as graphene (Gr) exhibit unique properties like high electrical conductivity, high surface to volume ratio, chemical and electrochemical stability and good mechanical strength.19,20 The high surface area of electrically conductive Gr sheets can give rise to high densities of attached analyte molecules. This in turn can facilitate high sensitivity and device miniaturization. Facile electron transfer between Gr and redox species opens up opportunities for sensing strategies based on direct electron transfer rather than mediation. It is not surprising, therefore, that Gr has recently attracted great attention worldwide from the electrochemical community. The production of Gr by the reduction of Gr oxide (GO) in chemical way produce hydroxyl (–OH) and carboxylate (–COOH) groups in the structure. These active functional groups enable the Gr structure to interact with metal NPs. These unique properties make Gr very useful for supporting metal NPs, and the obtained metal-NPs/Gr nanocomposites exhibit the synergistic effects and good sensitivity in their electrochemical detection behavior. The catalytic activity of the metal-NPs upon the electron transfer process paves the way for the production of metal-NPs/Gr composite based electrochemical sensors.21,22 Among the metal oxides, cupric oxide (CuO), well-known material of p-type semiconductor, can be promising candidate due to low cost, abundant resources, non-toxicity, and easy preparation in various shapes of nanosized dimensions.23 It has been widely investigated as electrode material for rechargeable Li-ion batteries, gas sensors, photocatalyst, CO oxidation catalysts and solar energy conversion.23–26 Thus, combining unique properties of Gr with interesting properties of CuO nanoparticles in electrochemical sensors, as electrode modifiers, offers great advantages including, mass transport enhancement, higher sensitivities, lower detection limits and faster kinetics of electron transfer in electrochemical reactions.23,27 Some researchers have determined two analytes out of three simultaneously, especially AA with AC and AC with CA.5,6,28–33 With attention to their direct redox reactions take place at very similar potentials at bare electrodes, which results in rather poor selectivity and difficulty on the determination of concentration of each species, this is a problem since some therapeutic formulations use these three species together.34,35 Recently, Fernandes and coworkers have been reported an attractive method for the simultaneous determination of AC, CA and AA using a voltammetric sensor modified with N-doped carbon nanotubes functionalized with MnFe2O4 nanoparticles.35 The linearity ranges of AC, CA and AA are 1.0 to 1000, 1.0 to 1100 and 2.0 to 100 μmol L−1, with detection limit (3Sb/m) of 0.83, 0.83 and 1.8 μmol L−1, respectively. The proposed method has limited in trace analysis of analytes and the researchers did not apply the sensor for analyzing the analytes in real samples such as pharmaceutical formulations, blood serum and urine.35 Therefore we aim to exploit the synergistic effect of the catalytic activity of CuO nanoparticles; together with the high conductivity and surface area of Gr sheets to serve as a potential electrode material for the simultaneous detection of AC, CA and AA with desired figures of merit. Well resolved peaks for the three were obtained at sensor by cyclic voltammetry (CV) and differential pulse voltammetry (DPV).
Experimental
Chemicals and apparatus
All chemicals and reagents used in this work were of analytical grade and used as received. AA, AC and CA as target analytes, paraffin oil and graphite powder were used as composition of electrodes were purchased from Merck Company. Deionized distilled water (DDW) was used to prepare all the solutions. Phosphate buffer, Britton–Robinson (B–R) and acetate buffers and KNO3 solution were prepared in DDW and were tested as the supporting electrolytes. Pharmaceutical samples were purchased from local drug stores.The electrochemical experiment including CV and DPV were recorded with an Autolab electrochemical analyzer, Model PGSTAT 302 N potentiostat/galvanostat (Eco-Chemie, Netherlands). A conventional three electrode cell assembly consisting of a platinum wire as an auxiliary electrode, an Ag/AgCl electrode as a reference electrode and carbon paste electrode (CPE) (unmodified and modified) as working electrodes were used. The morphological characterizations of all electrodes have been examined by means of scanning electrochemical microscopy, SEM (SEM-EDX, Philips Netherland). X-ray powder diffraction (XRD, 38066 Riva, d/G.Via M. Misone, 11/D (TN) Italy) was employed to analyze the chemical components of the composites. Fourier transform infrared (FT-IR) spectra were recorded in the range of 400–4000 cm−1 on a PerkinElmer, spectrum 100, FT-IR spectrometer. X-ray photoelectron spectroscopy (XPS) performed on a VG Microtech. The pH-measurements were done with a Metrohm pH meter (model 713).
Preparation of CuO nanoparticles and CuO/Gr
Gr oxide was prepared from natural graphite based on Hummers' method.36 The CuO/Gr composite and CuO nanoparticles were prepared by a simple liquid method.37,38 In a typical synthesis, 0.005 mol Cu (CH3COO)2·5H2O was dissolved in 50 mL of DDW with violent magnetic stirring, then 80 mg of Gr dispersed in 10 mL of ethanol (95%) was added to the above solution. Ammonia solution (10 mL, 25 wt%) was added to the resulting suspension last. After being stirred for 1 h, the resulting mixture was refluxed for 2 h. Vigorous stirring was maintained throughout the entire process. After the reaction, the solution was cooled to room temperature. The product was separated by centrifugation, washed with deionized water and absolute alcohol three times, and then dried in a vacuum oven at 80 °C for 10 h. CuO nanoparticles was prepared by a similar process for comparison without added Gr.39Preparation of the electrode
CPE was prepared by thoroughly hand mixing of graphite powder with appropriate amount of paraffin oil in a mortar using a pestle (75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25, w/w%). A portion of the composite mixture was packed firmly a piston-driven carbon paste electrode holder. The CuO/CPE was prepared by mixing 10% (w/w) CuO, 65% (w/w) graphite powder and 25% (w/w) paraffin oil in a mortar and pestle. The CuO–Gr/CPE was prepared by mixing the unmodified mixture with 15% w/w CuO–Gr and transferred into the CPE holder. A copper wire was inserted through the composite end of the working electrode to establish electrical contact.
:25, w/w%). A portion of the composite mixture was packed firmly a piston-driven carbon paste electrode holder. The CuO/CPE was prepared by mixing 10% (w/w) CuO, 65% (w/w) graphite powder and 25% (w/w) paraffin oil in a mortar and pestle. The CuO–Gr/CPE was prepared by mixing the unmodified mixture with 15% w/w CuO–Gr and transferred into the CPE holder. A copper wire was inserted through the composite end of the working electrode to establish electrical contact.
Preparation of real samples
All experiments were performed in compliance with the relevant laws and institutional guidelines of Ethics Committee and written informed consents were obtained from all volunteers. Different real samples containing analyte species were used to evaluate the analytical performance of the proposed sensor. Ten tablets were finely powdered in a mortar and certain amount of this powder dissolved in B–R solution pH 3.5 to prepare the stock solutions.Fresh serum and urine samples were originally obtained from two non-smoking patients (patient 1: male, 28 years, 96 kg, 178 cm, patient 2: male, 30 years, 85 kg, 180 cm). The serum was centrifuged and then after filtering, diluted 30 times with B–R buffer solution (pH = 3.5) without any further treatment. 10 mL of the fresh urine sample was centrifuged and the supernatant was filtered using a filter and then diluted with B–R buffer solution. The solution was transferred into the voltammetric cell to be analyzed without any further pretreatment. In order to reduce the matrix effect the standard additions method was employed in direct analysis of pharmaceutical samples.
Results and discussion
Surface characterization
The morphology of the typical products were investigated by SEM. Fig. 1a shows the SEM images of the Gr, CuO nanoparticles and CuO–Gr. It could be clearly seen that the Gr sheet exhibited a typical rippled and crumpled morphology and paper-like structure with single or very thin layers. From Fig. 1a, it was clear that the CuO nanoparticles were essentially fine and had a mean diameter of about 30 nm. The surfaces of the CuO–Gr become much rougher than pure Gr, indicating the growth of metal oxide nanoparticles on the surfaces. These particles are densely and homogeneously deposited onto the Gr sheets. Also, EDX spectrum of CuO indicates the high purity of the synthesized material where only Cu and O are present (Fig. 1b). The XRD patterns of the obtained Gr, CuO nanoparticles, CuO–Gr are shown in Fig. 1c. For XRD patterns of the CuO nanoparticles, all peaks can be confirmed to be the monoclinic phase of CuO (JCPDS no. 48-1548) with high crystallinity. No impurity peaks of other copper oxides were observed, which indicates the high purity of CuO nanoparticles. XRD of the synthesized Gr displayed a typical characteristic peak (002) at 2θ = 24.5° with d-spacing of 3.6 Å, which matches well with the reported literature for the formation of Gr.20 Also this results were confirmed by XPS analysis (Fig. S1†).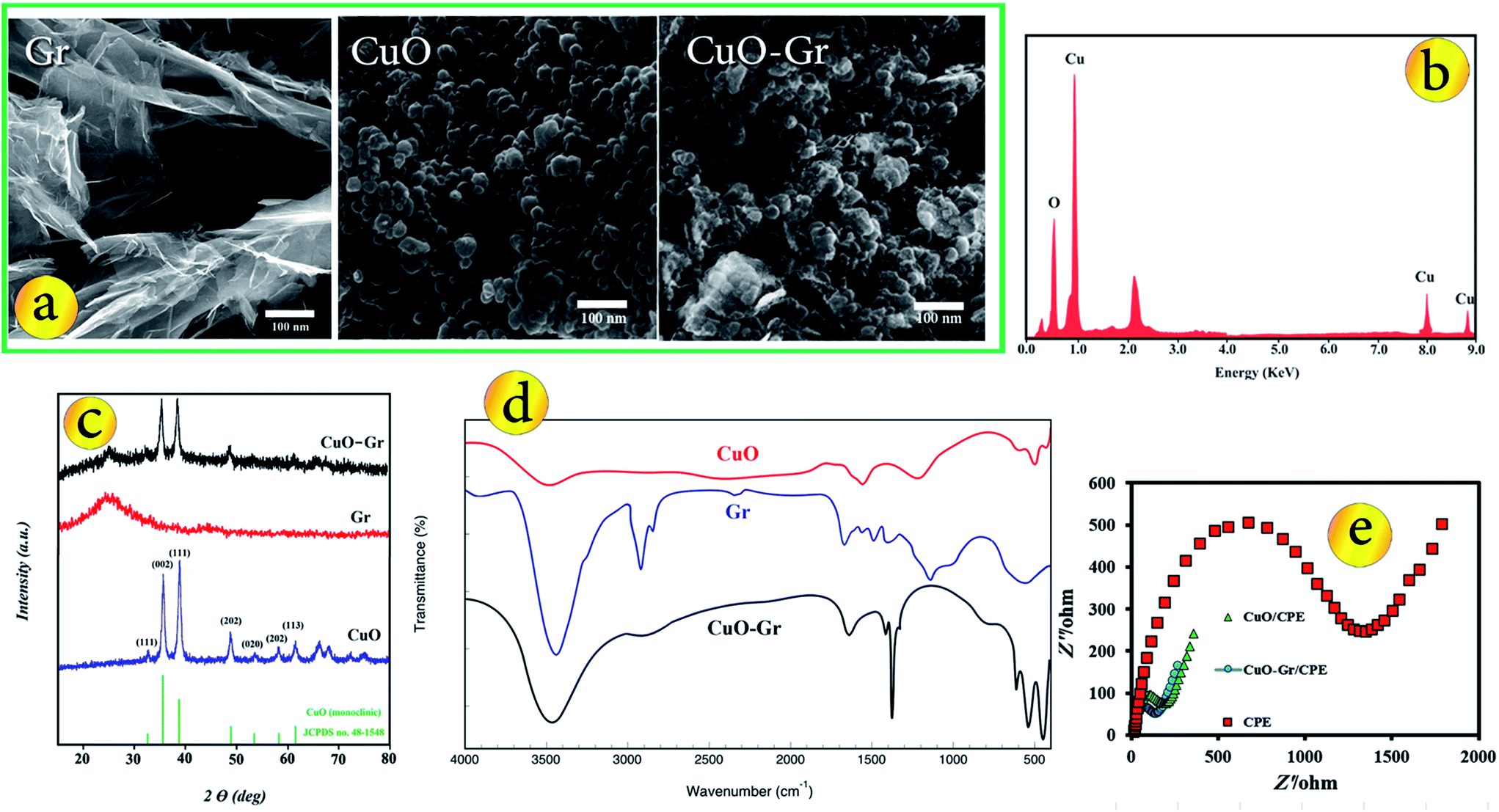 | ||
| Fig. 1 (a) SEM images from surface of Gr, CuO nanoparticles and CuO–Gr composite, (b) SEM-EDX spectrum of CuO nanoparticles, (c) XRD patterns of CuO nanoparticles, Gr and CuO–Gr composite and FT-IR spectra for CuO nanoparticles, Gr and CuO–Gr composite (d) and Nyquist plots for various electrodes (e). | ||
As can be seen, in the XRD pattern of CuO–Gr, the positions of diffraction peaks matched well with standard CuO and Gr. The pattern of CuO–Gr displayed obvious diffraction peaks of CuO nanoparticles, and the peak positions and relative intensities match well with the standard XRD data for CuO nanoparticles and the XRD pattern of the CuO–Gr shows an additional peak at 2θ = 24.5° attributed to the plane of the hexagonal graphite structure, suggesting that prepared composite composed of pure crystalline CuO nanoparticles were successfully decorated onto the Gr sheets.
The FT-IR spectra of the Gr, CuO nanoparticles and CuO–Gr composite (Fig. 1d) were also employed to confirm the chemical structure of prepared materials. The peak about 500 and 600 cm−1 related to the Cu–O stretching40 and the band around 3500 cm−1 corresponds to the stretching vibration of adsorbed water. FT-IR spectrum of CuO–Gr shows the stretching vibrations of C–O, C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C in 1076, 1352 and 1624 cm−1 respectively, indicate that a CuO–Gr composite is obtained.41
C in 1076, 1352 and 1624 cm−1 respectively, indicate that a CuO–Gr composite is obtained.41
Electrochemical impedance spectroscopy
Fig. 1e exhibits the Nyquist plots of the CPE, CuO/CPE and CuO–Gr/CPE. The CPE revealed a very large semicircle domain, implying a very high electron transfer resistance (Ret) of the redox probe. After the CPE was modified with CuO, the Ret was a small semicircle domain, showing that the CuO/CPE promoted conductivity. When the electrode was conjugated with CuO–Gr, the Ret slightly decreased. This was attributed to that the coating of Gr surface with CuO nanoparticles increased the ability of the redox probe to electron transfer. It can be seen that the electron transfer resistance decreases in the order of: bare CuO–Gr/CPE < CuO/CPE < CPE, which implying that CuO and Gr were excellent electric conducting materials and accelerated the electron transfer.Surface area study
The effective surface area of the electrodes can be determined by ferricyanide cyclic voltammetry (Fig. S2†) and the Randles–Sevcik equation.| Ip = (2.69 × 105)n3/2AC*D1/2ν1/2 | (1) |
Electrochemical behavior of analytes at unmodified and modified electrodes
In order to explain the electrochemical properties of modified electrode, the CV and DP voltammograms of 5 μmol L−1 AA, AC and CA on the surface of various electrodes including bare CPE, CuO/CPE and CuO–Gr/CPE in B–R buffer solution (pH = 3.5) were obtained (Fig. 2). In Fig. 2a–c can be seen, electrochemical oxidations of AA, AC and CA at bare CPE show a relatively weak and irreversible peak in 0.44, 0.529 and 1.494 V vs. Ag/AgCl. No anodic peak was observed within the investigated potential range on CuO/CPE and CuO–Gr/CPE in the absence of analytes. At CuO/CPE in the presence of analytes, the corresponding anodic peak current of every one of analytes increased in its peak current in comparison with CPE. When Gr was added to electrode composition, anodic peak current of the analytes have a remarkably increased in comparison with CuO/CPE and CPE.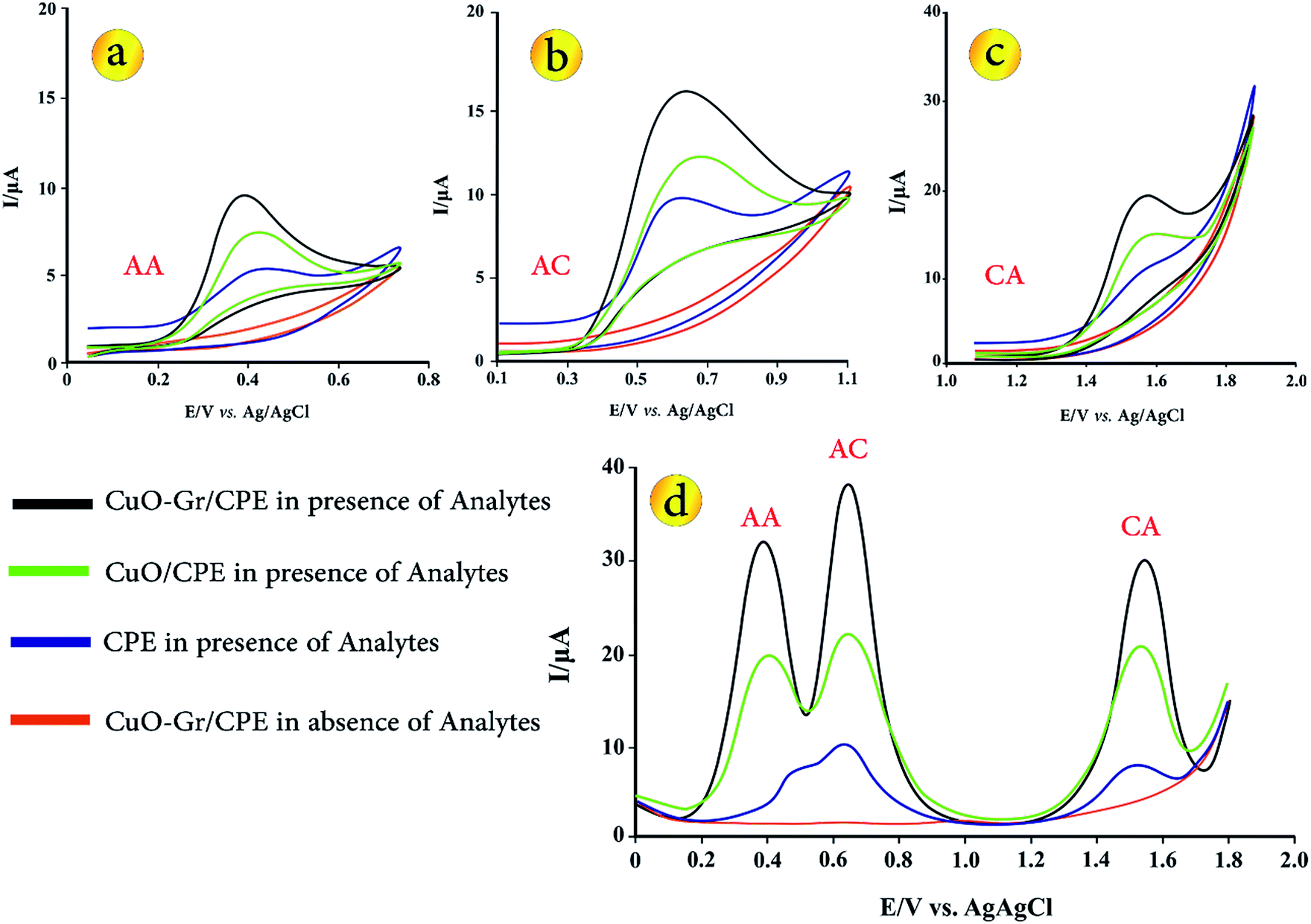 | ||
| Fig. 2 CVs for (a) 5.0 μmol L−1 AA, (b) 5.0 μmol L−1 AC, (c) 5.0 μmol L−1 CA and (d) DPVs of 5.0 μmol L−1 of AA, Ac and CA solution on the surface of various sensors (d). | ||
Fig. 2d presents the DPVs of different electrodes in B–R buffer solution (pH = 3.5) with 5.0 μmol L−1 AA, AC and CA. At the bare CPE in the presence of analytes, the oxidation peaks of AC and AA are not obviously separated. Due to peak potential values of analytes (0.505, 0.632 and 1.510 V vs. Ag/AgCl for AA, AC and CA), peaks separation between AA-CA and AC-CA are 1.005 and 0.878 V vs. Ag/AgCl, but oxidation peaks of AA and AC were strongly overlapped, thus simultaneous determination of analytes is not possible.
After adding of CuO in sensing layer (CuO/CPE), three separated and well defined peaks with increased peak currents were appeared. Moreover, CuO–Gr/CPE shows three distinct peaks for AA, AC and CA (∼0.387, 0.645 and 1.543 V) that can well be used for simultaneous determination of these biomolecules. The observations on CuO/CPE and CuO–Gr/CPE demonstrate that a negative shift with much enhanced anodic peak currents in comparison with CPE due to strong enhancement in the electron transfer rates of AA, AC and CA is taking place. Also, the apparent peak shapes for AA, AC and CA at modified electrodes are improved against those at CPE, so that the well-shaped peaks of these species can be observed with the presence of CuO–Gr providing an excellent electrochemical reactivity and increasing in the surface area of the electrodes. Moreover, no fouling was observed due to oxidation of the analytes.
Effect of pH
The electrochemical behaviour of the biological molecules are influenced by the nature of the electrolyte solution and pH. In order to evaluate the effect of different media on the sensor response, B–R buffer, phosphate buffer, acetate buffer and KNO3 solutions were tested and the analytical parameters in these media were obtained. In all buffer solution, simultaneous determination was possible because the anodic peak potentials for three species are well separated but the best results were obtained with the B–R buffer solution. Also, the peak currents of these biological compounds in B–R buffer solution were higher than those in other buffer solution. So this buffer was chosen for next studies.Because the proton takes part in the electrode reactions process of AA, AC and CA, the effect of the pH value on the voltammetric behaviour, the oxidation peak potentials and peak currents of AA, AC and CA at the CuO–Gr/CPE was investigated by DPV in a pH range of 2.5–8.5. It is clear that the oxidation peak potentials of the three molecules shift to negative values with the increase of pH values (Fig. 3a and c). The relationship between peak potential and pH is linear. The linear regression equations of AA, AC and CA are expressed as follows respectively:
| E = −0.0493pH + 0.6154, R2 = 0.9919 | (2) |
| E = −0.0604pH + 0.9134, R2 = 0.9934 | (3) |
| E = −0.0532pH + 1.7155, R2 = 0.9772 | (4) |
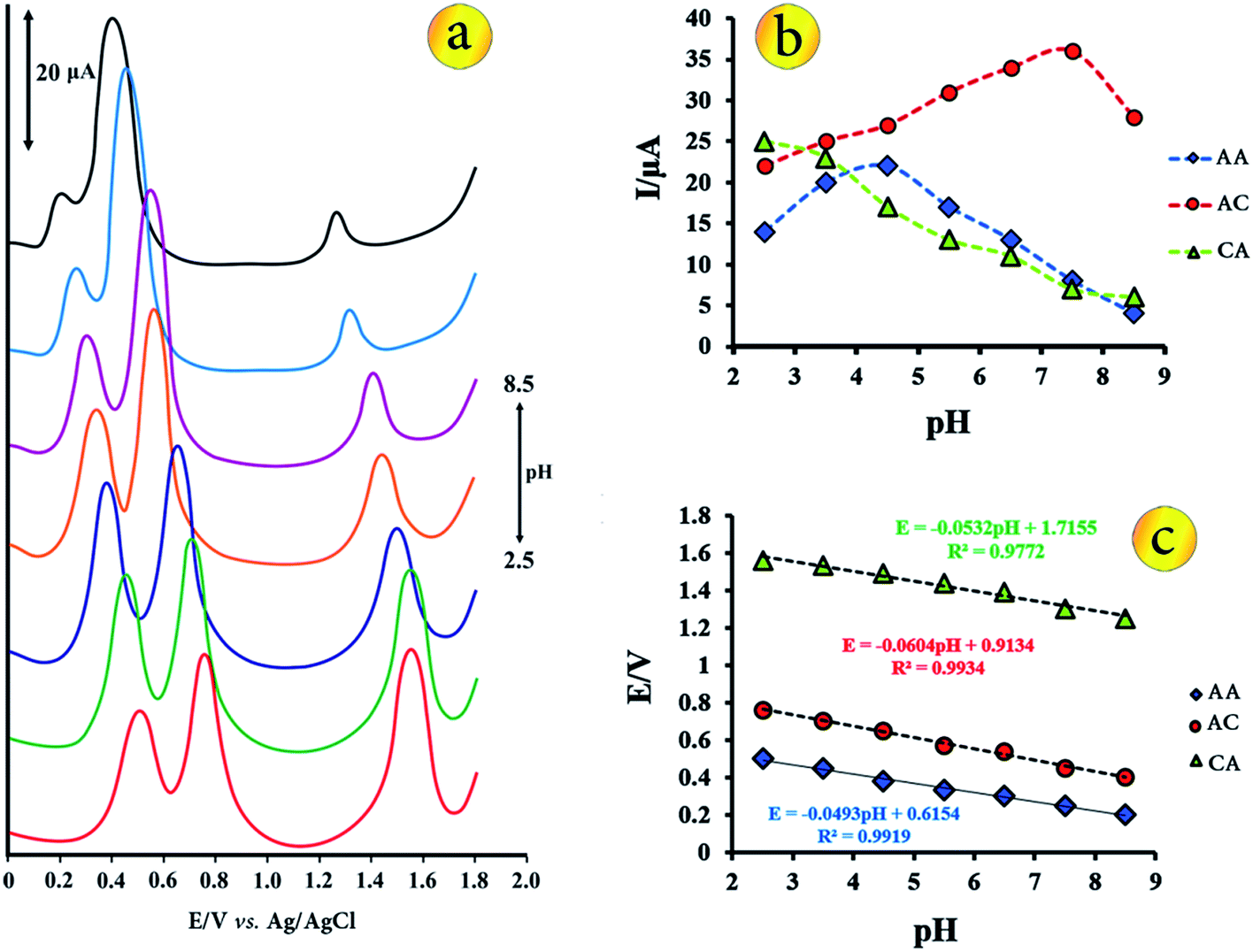 | ||
| Fig. 3 (a) 3D plots of influence of buffer pH on peak currents and potentials of analyte species, (b) plots of I vs. pH and (c) plots of Ep vs. pH from DP voltammograms of 5.0 μmol L−1 AA, AC and CA in different buffer solutions. | ||
These slopes are closed to the theoretical value of −59 mV pH−1 at 25 °C expected from the Nernst equation, indicates that electrochemical processes of each molecule involving the same number of protons and electrons.
The changes of the peak currents of oxidation of the biological molecules with pH recorded in Fig. 3b. Within the pH ranges of 2.5 to 8.5, the anodic peak current of AA oxidation increased gradually from 2.5 to 4.5 and then decreased to pH 8.5. In the case of AC, the peak current increased from 2.5 to 7.5 and then decreased to pH 8.5. The oxidation current of CA is maximum value in pH = 2.5 and decreased to pH = 8.5. In finally, considering the separation peaks, peak currents and the detection sensitivity, the buffer solution pH of 3.5 was chosen as the optimal pH for the simultaneous determination of AC, CA and AA.
Effect of scan rate on the electrochemical oxidation of AA, AC and CA
The influence of scan rate on the electrochemical response of 5.0 μmol L−1 of AA, AC and CA at the CuO–Gr/CPE was investigated by CV in the scan rate range from 50 to 350 mV s−1 in B–R buffer solution with pH 3.5 (Fig. 4). The results showed that the anodic peak current (Ipa) was increased gradually linearly with the increase of square root rates (ν1/2), indicating a diffusion controlled electrode process. The linear regression equations of AA, AC and CA are expressed as follows respectively:| Ipa = 1.0636ν1/2 − 1.2062, R2 = 0.9963 | (5) |
| Ipa = 1.4481ν1/2 − 0.2862, R2 = 0.9899 | (6) |
| Ipa = 1.1457ν1/2 − 0.8117, R2 = 0.9908 | (7) |
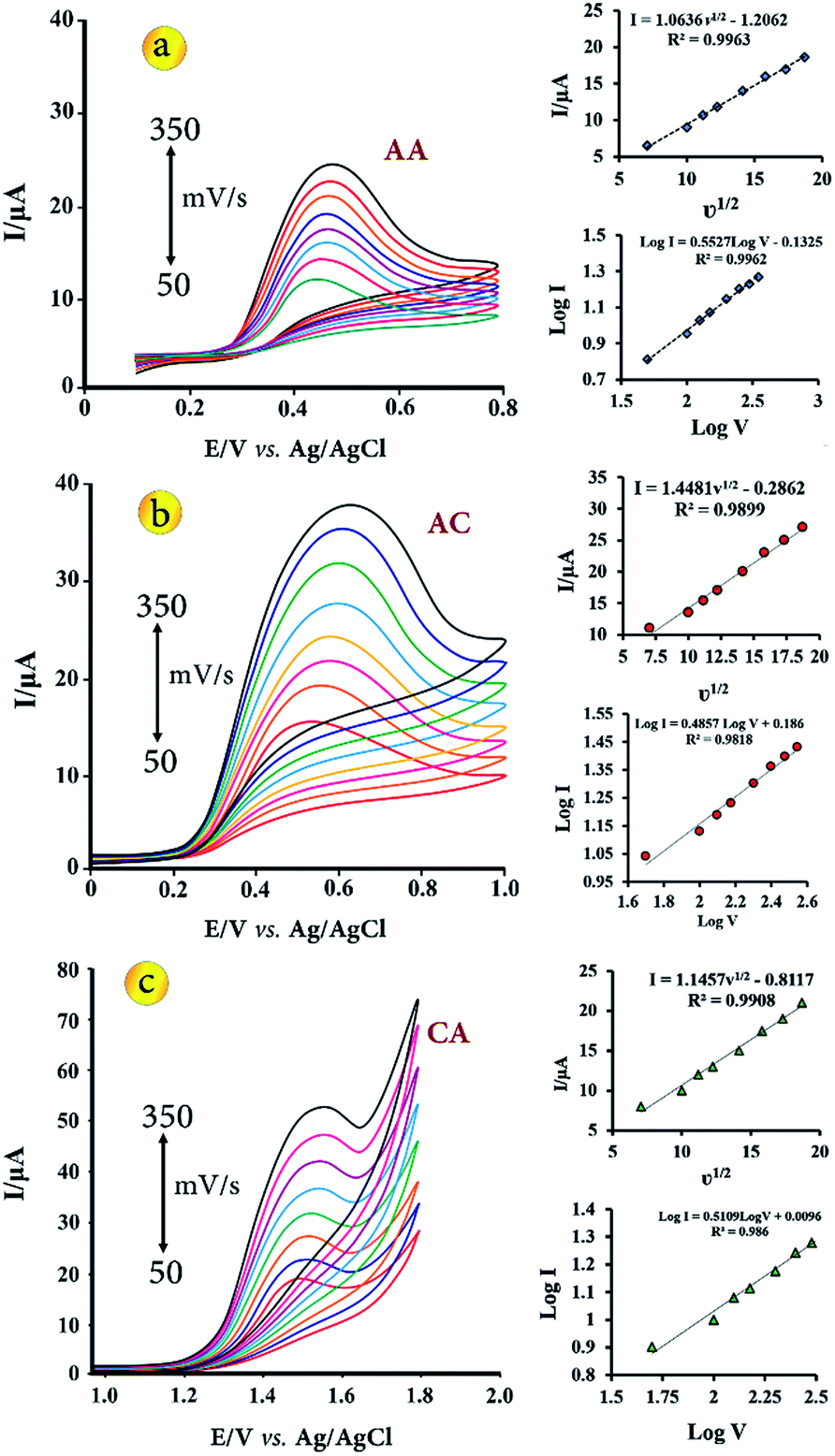 | ||
| Fig. 4 CVs for CuO–Gr/CPE (in B–R buffer solution with pH 3.5) containing 5.0 μmol L−1 of AA (a), AC (b) and CA (c) with scan rates ranging from 50 to 350 mV s−1, respectively. Insets show the linear relationship of the peak current vs. square root of the scan rate (ν1/2) and log peak currents vs. log scan rate. | ||
Also, relationship between logIpa and logν was investigated. Linear regression equations of AA, AC and CA as logIpa = 0.5527logν − 0.1325 (R2 = 0.9962), Ipa = 0.4857logν + 0.186 (R2 = 0.9818) and logIpa = 0.5109logν + 0.0096 (R2 = 0.986), respectively. According to the slope values, which were equal about 0.5, the electrochemical processes are diffusion-controlled.
Analytical performance, stability and reproducibility
Under the optimal conditions that described in the previous sections, calibration plots for the simultaneous determination of AA, AC and CA at the modified electrode were obtained by DPV (Fig. 5). For the simultaneous determination of target species, keeping the concentration of one species constant at 1.50 μmol L−1 while changing that of the other from 0.025 to 5.30 μmol L−1 (Fig. 5a–c). As shown in Fig. 5a–c, it can be seen that the oxidation peak current of each of the molecules, increased linearly with the increasing concentrations in the range from 0.025 μmol L−1 to 5.30 μmol L−1 in the presence of two other molecules at a concentration of 1.50 μmol L−1. The linear regression equation for AA, AC and CA were Ipa = 4.487CAA + 0.4662 (R2 = 0.998), Ipa = 5.5774CAC + 0.7089 (R2 = 0.999) and Ipa = 4.863CCA + 0.6947 (R2 = 0.999), simultaneously.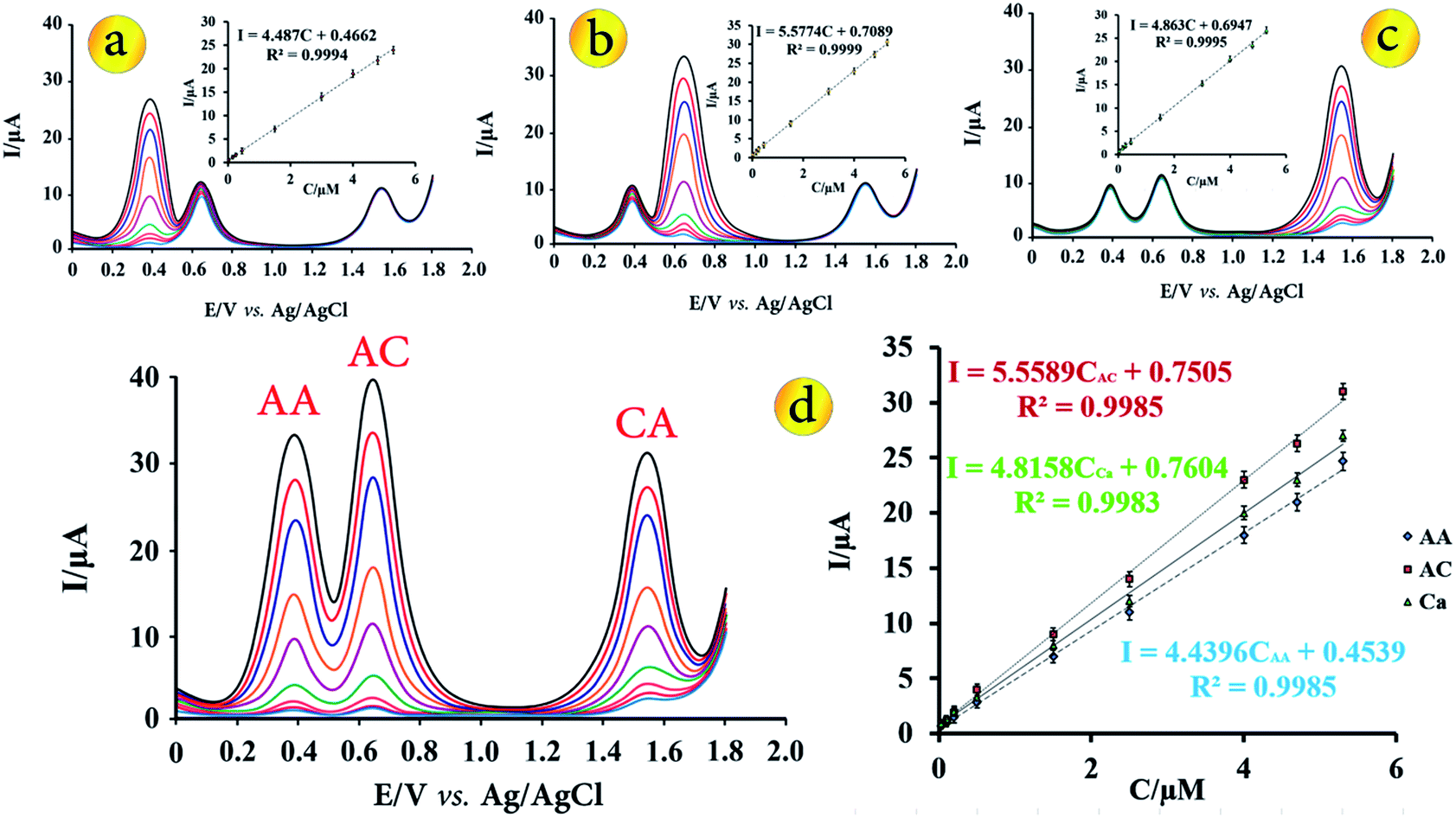 | ||
| Fig. 5 DP voltammograms using CuO–Gr/CPE under optimum conditions in solution containing different concentrations of AA in the presence constant concentrations of AC and CA (a). Inset shows calibration plot of the peak currents as a function of analyte concentrations. (b) is related to AC in the presence constant concentrations of AA and CA, (c) is related to CA in the presence constant concentration of AA and AC, respectively. (d) DPV for different concentrations of AA, AC and CA under optimum conditions. | ||
Fig. 5d shows the DPVs obtained at modified electrode for different concentrations of AA, AC and CA in B–R buffer solution with pH = 3.5. The anodic currents of AA, AC and CA increased linearly with increasing their concentrations over the range of 0.025 to 5.30 μmol L−1 with the linear regression equation of Ipa = 4.4396CAA + 0.4539 (R2 = 0.9985), Ipa = 5.5589CAC + 0.7505 (R2 = 0.9985) and Ipa = 4.8158CCA + 0.7604 (R2 = 0.9983), respectively. The slope of the regression equations for the calibration graph of each species is nearly equal to that without the other species, indicating that they do not interfere in the determination of each other. Based on the 3Sb/m, which Sb is standard deviation of blank determinations and m is slope of calibration plot, the detection limit for determination of AA, AC and CA were found to be 0.011, 0.008 and 0.010 μmol L−1, respectively.
The repeatability, reproducibility and stability of CuO–Gr/CPE were investigated in the B–R buffer solution containing 1.00 μmol L−1 of each species. According to successive measurements in 15 times, the proposed sensor showed an acceptable repeatability with a relative standard deviation (RSD) of 2.45%, 2.67% and 2.38% for the oxidation peak currents of AA, AC and CA, respectively. RSD values 3.5%, 3.8% and 3.7% for AA, AC and CA were obtained respectively, with ten sensors prepared independently using the same procedure. The modified electrode was stored in ambient at lab for 30 days. After 10, 15, 20 and 30 day, the anodic peak currents of the sensor, retained more than 99.1%, 98.4%, 96.3% and 92.2% compared with initial value which shows good stability of electrode for the analysis of real samples.
The comparison of CuO–Gr/CPE with other modified electrodes for the determination of AA, AC and AA was listed in Table 1. From Table 1, it could be seen that CuO–Gr/CPE had the comparable sensitivities and detection limits for the detection of AA, AC and CA.
| Electrode | Method | Linear range (μmol L−1) | Detection limit (μmol L−1) | Refs. no | ||||
|---|---|---|---|---|---|---|---|---|
| AC | AA | CA | AC | AA | CA | |||
| MWCNTs/GCE | SWV | — | 10–500 | 10–500 | — | 0.01 | 0.00352 | 5 |
| Boron-doped diamond electrode | DPV | 0.5–83 | — | 0.5–83 | 0.49 | — | 0.035 | 7 |
| Flavonoid nanostructured/GCE | DPV | 0.9–80 | — | 10–110 | 0.78 | — | 3.54 | 28 |
| Carbon nanotubes/carbon–ceramic electrode | DPV | 0.08–200 | — | 0.41–300 | 0.05 | — | 0.29 | 29 |
| MWCNTs dispersed in polyhistidine/GCE | DPV | 0.25–10 | 25–2500 | — | 0.032 | 0.76 | — | 30 |
| Boron-doped diamond film electrode | DPV | — | — | 1–1000 | — | — | 0.23 | 31 |
| SWCNTs/carbon–ceramic electrode | DPV | 0.2–150 | 5–700 | — | 0.12 | 3 | — | 32 |
| Gold–silver bimetallic nanotubes in a chitosan matrix/GCE | Amperometry | — | 5–2000 | — | — | 2 | — | 33 |
| GCE | DPV | 0–0.36 | 0–0.2 | 0–0.26 | 0.048 | 0.05 | 0.043 | 34 |
| MnFe2O4@CNT-N/GCE | 1–1000 | 2–100 | 1–1100 | 0.83 | 1.8 | 0.83 | 35 | |
| CuO–Gr/CPE | DPV | 0.025–5.30 | 0.025–5.30 | 0.025–5.30 | 0.008 | 0.011 | 0.010 | This work |
Interferences study
The interferences of the other substances on response of sensor and selectivity of the modified electrode for the determination of AA, AC and CA was studied under the optimum conditions with 1.00 μmol L−1 of each target analytes at pH = 3.5. Substances that commonly found with the target analytes in pharmaceuticals or in biological fluids were chosen that were include agents Na+, Mg2+, Ca2+, NO3−, SO42−, SO32−, CO32−, fructose, lactose, glucose, glutathione, N-acetyl-L-cysteine, dopamine, urea and thiourea. Tolerance limit was taken as the maximum concentration of foreign substances that caused an approximate relative error of ±5%. The absence of significant shift in the peak currents recorded in the presence of the interfering species proved that CuO–Gr/CPE can be considered as a good electrochemical sensor for recognition of AA, AC and CA in aqueous solutions.Application of method to real samples
To confirm the usefulness of CuO–Gr/CPE at optimum conditions by DPV method, the proposed method was successfully applied to the simultaneous determination of AA, AC and CA in real samples with different matrixes. Prepared real samples were diluted to the same volume by B–R buffer solution at pH 3.5. Each of these solutions was transferred into the electrochemical cell and the voltammetric measurements were performed using the standard addition method. The results of simultaneous determinations of target analytes by purposed DPV method and HPLC (for comparison) reported in Tables 2 and 3. The recoveries of the samples indicating that there were no important matrix interferences for the samples analysed by the proposed DPV method. They were acceptable and confirmed the applicability of the modified electrode for AA, AC and CA measurements in real samples matrixes.| Sample | Analyte | Added | Found | Recovery (%) | HPLC method |
|---|---|---|---|---|---|
| a Not detected. | |||||
| Urine 1 | AA | 0.00 | 0.00 | — | NDa |
| 2.00 | 2.06 ± 0.07 | 103 | 2.01 ± 0.01 | ||
| AC | 0.00 | 0.00 | |||
| 2.00 | 2.07 ± 0.07 | 103.5 | 2.03 ± 0.02 | ||
| CA | 0.00 | 0.00 | |||
| 2.00 | 1.96 ± 0.04 | 98 | 2.02 ± 0.04 | ||
| Urine 2 | AA | 0.00 | 0.00 | — | ND |
| 2.00 | 2.04 ± 0.05 | 102 | 2.03 ± 0.03 | ||
| AC | 0.00 | 0.00 | 0.00 | ||
| 2.00 | 2.02 ± 0.04 | 101 | 1.98 ± 0.02 | ||
| CA | 0.00 | 0.53 ± 0.07 | — | 0.55 ± 0.01 | |
| 2.00 | 2.46 ± 0.06 | 96.5 | 2.54 ± 0.02 | ||
| Serum | AA | 0.00 | 5.06 ± 0.07 | — | 5.08 ± 0.02 |
| 2.00 | 7.02 ± 0.05 | 98 | 7.09 ± 0.03 | ||
| AC | 0.00 | 0.00 | — | ND | |
| 2.00 | 2.02 ± 0.06 | 101 | 1.96 ± 0.03 | ||
| CA | 0.00 | 0.00 | — | ND | |
| 2.00 | 1.95 ± 0.05 | 97.5 | 2.01 ± 0.02 | ||
| Sample | Analyte | Tablet label values (mg) | Found (mg) | HPLC value (mg) |
|---|---|---|---|---|
| Tablet 1 (Rahafan©) | AC | 325 | 322 ± 3 | 323 ± 2 |
| CA | 40 | 41 ± 2 | 41 ± 3 | |
| Tablet 2 (EXCEDRIN©) | AC | 250 | 252 ± 3 | 250 ± 1 |
| CA | 65 | 63 ± 5 | 66 ± 2 | |
| Tablet vitamin C | AA | 500 | 492 ± 4 | 498 ± 3 |
Conclusions
Monitoring the levels of AA, AC and CA is of great importance and has always been the target of researchers. In this paper for the first time, simultaneous determination of AA, AC and CA based on DPV at CuO–Gr/CPE was studied. The best responses of the sensor were achieved in B–R buffer solution pH 3.5. Due to facile electron transfer reaction and high selectivity at sensing layer, this sensor separates the voltammetric signals of the three analytes with enhanced peak currents compared to other reported sensor. The detection limits were found to be 0.011, 0.008 and 0.010 μmol L−1 for AA, AC and CA, respectively. Compared with other electrodes (shown in Table 1), the proposed electrode shows excellent advantages such as wide linear ranges and low detection limits. The interfering study of some biological species and common inorganic ions showed no interference with the selective determination of target molecules. The sensor was successfully applied for simultaneous determination of AA, AC and CA in various real samples with different matrix. According to data that obtained from real samples analysis with the proposed method and comparison with HPLC, the sensor could be applied to the determination of AA, AC and CA in real pharmaceutical and biological samples with promising results.Acknowledgements
This research was supported by the Research Council of the Payame Noor University.References
- A. Afkhami, H. Khoshsafar, H. Bagheri and T. Madrakian, Anal. Chim. Acta, 2014, 831, 50–59 CrossRef CAS PubMed.
- A. A. Ensafi, H. Karimi-Maleh and S. Mallakpour, Electroanalysis, 2012, 24, 666–675 CrossRef CAS.
- A. J. Jeevagan and S. A. John, Electrochim. Acta, 2012, 77, 137–142 CrossRef CAS.
- A. Krisko, M. Kveder and G. Pifat, Clin. Chim. Acta, 2005, 355, 47–53 CrossRef CAS PubMed.
- V. K. Gupta, A. K. Jain and S. K. Shoora, Electrochim. Acta, 2013, 93, 248–253 CrossRef CAS.
- S. Kerrigan and T. Lindsey, Forensic Sci. Int., 2005, 153, 67–69 CrossRef CAS PubMed.
- B. C. Lourencao, R. A. Medeiros, R. C. Rocha-Filho, L. H. Mazo and O. Fatibello-Filho, Talanta, 2009, 78, 748–752 CrossRef CAS PubMed.
- S. Shahrokhian and E. Asadin, Electrochim. Acta, 2010, 55, 666–672 CrossRef CAS.
- C. Q. Wang, J. Du, H. W. Wang, C. Zou, F. X. Jiang, P. Yang and Y. K. Du, Sens. Actuators, B, 2014, 204, 302–309 CrossRef CAS.
- C. Zhou, S. Li, W. Zhu, H. Pang and H. Ma, Electrochim. Acta, 2013, 113, 454–463 CrossRef CAS.
- P. Koblova, H. Sklenarova, I. Brabcova and P. Solich, Anal. Methods, 2012, 4, 1588–1591 RSC.
- A. Bozdoǧan, A. M. Acar and G. K. Kunt, Talanta, 1992, 39, 977–979 CrossRef.
- M. Blanco and M. Alcala, Eur. J. Pharm. Sci., 2006, 27, 280–286 CrossRef CAS PubMed.
- H. Bagheri, A. Afhami, P. Hashemi and M. Ghanei, RSC Adv., 2015, 5, 21659–21669 RSC.
- H. Bagheri, R. P. Talemi and A. Afkhami, RSC Adv., 2015, 5, 58491–58498 RSC.
- H. Bagheri, A. Shirzadmehr and M. Rezaei, J. Mol. Liq., 2015, 212, 96–102 CrossRef CAS.
- H. Bagheri, S. M. Arab, H. Khoshsafar and A. Afkhami, New J. Chem., 2015, 39, 3875–3881 RSC.
- H. Bagheri, A. Afkhami, H. Khoshsafar, M. Rezaei, S. J. Sabounchei and M. Sarlakifar, Anal. Chim. Acta, 2015, 870, 56–66 CrossRef CAS PubMed.
- A. Afkhami, H. Khoshsafar, H. Bagheri and T. Madrakian, Sens. Actuators, B, 2014, 203, 909–918 CrossRef CAS.
- H. Bagheri, A. Afkhami, Y. Panahi, H. Khoshsafar and A. Shirzadmehr, Mater. Sci. Eng., C, 2014, 37, 264–270 CrossRef CAS PubMed.
- E. Er, H. Çelikkan, N. Erk and M. L. Aksu, Electrochim. Acta, 2015, 157, 252–257 CrossRef CAS.
- S. Pruneanu, F. Pogacean, A. R. Biris, M. Coros, F. Watanabe, E. Dervishi and A. S. Biris, Electrochim. Acta, 2013, 89, 246–252 CrossRef CAS.
- A. Pendashteh, M. F. Mousavi and M. Safi Rahmanifar, Electrochim. Acta, 2013, 88, 347–357 CrossRef CAS.
- B. Wang, X. L. Wu, C. Y. Shu, Y. G. Guo and C. R. Wang, J. Mater. Chem., 2010, 20, 10661–10664 RSC.
- S. Liu, J. Q. Tian, L. Wang, Y. L. Luo and X. I. Sun, Catal. Sci. Technol., 2012, 2, 339–344 CAS.
- J. Q. Tian, H. Y. Li, Z. C. Xing, L. Wang, Y. L. Luo, A. M. Asiri, A. R. O. Al-Youbi and X. P. Sun, Catal. Sci. Technol., 2012, 2, 2227–2230 CAS.
- L. Q. Luo, L. M. Zhu and Z. X. Wang, Bioelectrochemistry, 2012, 88, 156–163 CrossRef CAS PubMed.
- M. Amiri-Aref, J. B. Raoof and R. Ojani, Sens. Actuators, B, 2014, 192, 634–641 CrossRef CAS.
- B. Habibi, M. Jahanbakhshi and M. Abazari, J. Iran. Chem. Soc., 2014, 11, 511–521 CrossRef CAS.
- P. R. Dalmasso, M. L. Pedano and G. A. Rivas, Sens. Actuators, B, 2012, 173, 732–736 CrossRef CAS.
- E. O. Faria, A. C. V. L. Junior, D. E. P. Souto, F. R. F. Leite, F. S. Damos, R. C. S. Luz, A. S. dos Santos, D. L. Franco and W. T. P. Santos, Electroanalysis, 2012, 24, 1141–1146 CrossRef CAS.
- B. Habibi, M. Jahanbakhshi and M. H. Pournaghi-Azar, Anal. Biochem., 2011, 411, 167–175 CrossRef CAS PubMed.
- P. H. Yang, X. Gao, L. Wang, Q. Wu, Z. C. Chen and X. F. Lin, Microchim. Acta, 2014, 181, 231–238 CrossRef CAS.
- O. W. Lau, S. F. Luk and Y. M. Cheung, Analyst, 1989, 114, 1047–1051 RSC.
- D. M. Fernandes, N. Silva, C. Pereira, C. Moura, J. M. C. S. Magalhães, B. Bachiller-Baeza, I. Rodríguez-Ramos, A. Guerrero-Ruiz, C. Delerue-Matos and C. Freire, Sens. Actuators, B, 2015, 218, 128–136 CrossRef CAS.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- C. W. Sun, J. Sun, G. L. Xiao, H. R. Zhang, X. P. Qiu, H. Li and L. Q. Chen, J. Phys. Chem. B, 2006, 110, 13445 CrossRef CAS PubMed.
- S. F. Zheng, J. S. Hu, L. S. Zhong, W. G. Song, L. J. Wan and Y. G. Guo, Chem. Mater., 2008, 20, 3617 CrossRef CAS.
- B. Wang, X. Long Wu, C. Shu, Y. Guo and C. Wang, J. Mater. Chem., 2010, 20, 10661–10664 RSC.
- K. Kliche and Z. V. Popovic, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 42, 10060–10066 CrossRef.
- B. Zhao, P. Liu and Y. Jiang, J. Mater. Chem. A, 2013, 1, 367 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra14138a |
| This journal is © The Royal Society of Chemistry 2015 |