
Fractionation of carboxylate anions from aqueous solution using chitosan cross-linked sorbent materials
Abstract
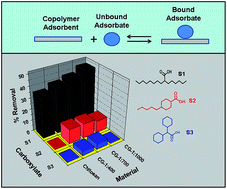
The sorptive uptake properties are reported for chitosan and cross-linked chitosan-glutaraldehyde (CG) polymers with single component carboxylic acids (surrogates; Si, i = 1–4). The single component surrogates are treated as model compounds to represent traditional naphthenic acids (NAs) with variable hydrogen deficiency (z) and carbon number (C), as follows: 2-hexyldecanoic acid (z = 0 and C = 16; S1), trans-4-pentylcyclohexanecarboxylic acid (z = −2 and C = 12; S2), dicyclohexylacetic acid (z = −4; C = 14; S3) and 2-ethyl hexanoic acid (z = 0 and C = 8; S4). Cross-linked polymers were synthesized at variable C : G mole ratios (i.e. 1 : 400, 1 : 700, and 1 : 1000) and characterized using FTIR and TGA. The uptake studies were evaluated at pH 6.00 and 9.00 in aqueous solution. Corresponding equilibrium uptake parameters were evaluated by several isotherm models (Sips, Freundlich, and Langmuir). The Freundlich model provided the “best-fit” for S1, S2 and S3. The Sips isotherm model gave the “best-fit” results for S4. The Freundlich sorption affinity constant (KF; L mg g−1) and the Sips monolayer adsorption capacity (Qm; mg g−1) for the sorbent systems was estimated. The parameters for each Si vary as the C : G mole ratio increases (KF, S1: 0.00800–6.65, S2: 1.41–2.45, S3: 1.29–2.93 and Qm, S4: 28.4–33.5). Differences in the uptake of Si species by the sorbents were attributed to steric effects between Si with ring systems (z < 0) versus linear alkyl fragments (z = 0). Fractionation of equimolar mixtures of Si reveal that the sorbents display unique and variable binding affinity with remarkable molecular selective uptake (given in parentheses; L mg g−1): S1 (4–30) > S2 (4–10) > S3 (3–9). The fractionation efficacy of Si species is attributed to molecular sieving effects due to the tunable micropore sites of the cross-linked polymer, in accordance with the variable cross-linker content of the chitosan framework. Regeneration studies revealed the utility of these materials in cyclic adsorption–desorption processes.
 Please wait while we load your content...
Please wait while we load your content...

