
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licencep-Nitrobenzenesulfonamides and their fluorescent dansylsulfonamides derived from N-alkylated o-(purine-methyl)anilines as novel antitumour agents†
Fátima
Morales
a,
Ana
Conejo-García
ab,
Alberto
Ramírez
c,
Cynthia
Morata
de,
Juan Antonio
Marchal
*de and
Joaquín M.
Campos
*ab
aDepartment of Pharmaceutical and Organic Chemistry, c/Campus de Cartuja s/n, Faculty of Pharmacy, University of Granada, 18081 Granada, Spain. E-mail: jmcampos@ugr.es; Fax: +34 958 24 38 45; Tel: +34 958 24 38 50
bBiosanitary Institute of Granada (ibs.GRANADA), SAS-Universidad de Granada, 18071 Granada, Spain
cDepartment of Health Sciences, Paraje de las Lagunillas s/n, University of Jaén, Jaén, Spain
dBiopathology and Medicine Regenerative Institute (IBIMER), University of Granada, Granada, Spain. E-mail: jmarchal@ugr.es; Fax: +34 958 24 62 96; Tel: +34 958 24 93 21
eBiosanitary Institute of Granada (ibs.GRANADA), 18071 Granada, Spain
First published on 3rd September 2015
Abstract
We have designed and synthesized a novel series of fourteen alkylated purines with notable in vitro anti-proliferative activities against three cancerous cell lines. In vivo studies show absorption by the oral route of administration with systemic distribution and low toxicity for the two more active derivatives.
Cancer is one of the most important health issues and the leading cause of death worldwide. Collaboration between researchers, epidemiologists and clinicians is essential to improve cancer control. Recent advances in the knowledge of cancer development have allowed researchers to determine that tumours are constituted by differentiated tumour cells and their precursor cells, also named as tumour-initiating cells or cancer stem cells (CSCs). These CSCs are a small tumour subpopulation responsible for initiating and maintaining tumour growth and are characterized by their self-renewal capacity and differentiation ability, the high metastatic potential and their quiescent state makes them resistant to chemo- and radio-therapy.1 Conventional anticancer therapies kill the rapidly proliferating bulk differentiated cancer cells but obviate the quiescent CSCs. Therefore, development of specific therapies targeted at CSCs holds hope to gain the fight against cancer.
We have previously described a series of cyclic and acyclic O,N-acetals (1 and 2, Fig. 1) as potent anti-tumour compounds.2–5 The IC50 values against the MCF-7 cell line were below 1 μM, 1 also called bozepinib, and 2 being the most potent structures so far reported (IC50 = 0.355 μM in both cases). Moreover, they are the most selective compounds against the MCF-7 cell line [TIs (Therapeutic Index) = 5.14 and 5.25, respectively] in relation to the normal one.2 cDNA microarray technology reveals potential drug targets, mainly centred on positive apoptosis regulatory pathway genes, and the repression of genes involved in carcinogenesis, proliferation, tumour invasion and CSCs related pathways.3 Bozepinib is a potent anti-tumour agent that induces apoptosis in breast and colon cancer cells mediated by the double-stranded RNA-dependent protein kinase (PKR).4 The combination with IFNα potentiates the apoptosis induced by bozepinib and also enhances autophagy and senescence,4 both processes of great importance in tumour cells that show resistance to conventional chemotherapy. Bozepinib is able to exert an inhibitory activity against the formation of both mamo- and colono-spheres and eliminates aldehyde dehydrogenase enriched (ALDH+) CSC subpopulations at a low micromolar range. Moreover, the inhibitory effect of bozepinib on CSC pathways was emphasized by the down-regulation of c-Myc, β-CATENIN and SOX2 proteins and the up-regulation of the hedgehog-signalling repressor form of GLI-3. Bozepinib also shows in vivo anti-tumour and anti-metastatic efficacy in xenotransplanted nude mice without presenting sub-acute toxicity.5
 | ||
| Fig. 1 The arrows and the legends on top of them show the corresponding structural modifications carried out for the design of the target molecules. | ||
In the present paper we present a new series of acyclic compounds with the aim to develop new anti-tumour agents based on the following modifications of the acyclic analogue of bozepinib (2), which in spite of being obtained as a by-product during the formation of bozepinib (1), its remarkable anti-proliferative properties2 justify to be considered as a new prototype: in short, the change of position of the purine base to the benzylic carbon and concomitant replacement of the O,N-acetalic function for an O,O-acetalic one, in the form of the 1,3-dioxolane group (Fig. 1). The anti-tumour activity has recently been described of a series of derivatives having a 1,3-dioxolane moiety in their structures (3). Such compounds are inhibitors of the proliferation and eradication of cancer cells, besides being inhibitors of CSCs.6 We maintain the p-nitrobenzenesulfonyl group and we introduce a linker between the sulfonamide and the dioxolane (n = 1, 2). We also include the 5-FU derivative (13) in order to act as a reference (Fig. 2).
 | ||
| Fig. 2 The fourteen molecules studied in this communication. | ||
The target compounds 4–13 (Fig. 2) are obtained following the synthetic procedure described in Scheme S1† (R1 = A; R2 = C, D, E and F). Compounds 18–20 were synthesized as previously described.7 When 21 and 22 are treated with tetrabutylammonium fluoride (TBAF) in anhydrous THF to give the de-protected compounds 25 and 26, by-products 29 and 30 are also obtained in smaller yields respectively (Scheme S2†). Compounds 29 and 30 are the result of a Smiles rearrangement,8 in which the p-nitrophenyl group migrates to the hydroxyl group through the formation of the mono-nitro spiro-complex (Scheme S2†).9 Target compounds 4–13 were obtained by microwave-assisted Mitsunobu reaction. Microwave-assisted organic synthesis is becoming instrumental for the rapid synthesis of drugs.10 The two following facts must be underlined: (a) reaction between 26 and adenine yields, apart from the N-9 derivative 11, the N-3 derivative 12;11 and (b) reaction between 27 (and 28) and 2,6-dichloropurine yields both the N-9 derivative 14 (and 15 when starting from 28), and the N-7 derivative 16 (and 17 when starting from 28).
The effects on cell proliferation were investigated on the human breast cancer cell line MCF-7, human colon carcinoma cell line HCT-116 and the human malignant melanoma cell line A-375. The anti-proliferative activities for 4–17 against the three tumour cell lines are shown in Table S1† in μM units. The three most promising compounds are 8, 9 and 10 (with IC50 values ranging from 0.134 μM against A-375, and 4.110 μM against HCT-116 for 10).
Fluorescence is a very useful technique to elucidate the mechanism of action of drugs inside the cell and to know their distribution in tissues and organs. Knowing drug distribution is critical for the design of more selective and efficient agents in cancer therapy, although this knowledge is neither always easy nor cheap. However, fluorescence technique provides a comprehensive tool for investigating these aspects in single cells and whole tissue.12 Some drugs are inherently auto-fluorescent and therefore can be tracked using microscopy techniques, such as topotecan and camptothecin13 but others need to be labelled with chromophores such as the ones shown in Fig. S1.† As an example, the mechanism of action of the anti-tumour drug paclitaxel by the stabilization of the microtubules was disclosed by the fluorescence techniques using fluorescein as a label in the drug.14 We have chosen the p-nitrobenzene sulfonyl derivative with n = 2 and the 2,6-dichloropurine moiety (10), because it is the most active compound against the A-375 cell line, in order to study the in vitro and in vivo drug distribution by fluorescence. Epidemiological studies showed that the incidence of melanoma is increasing at a rate faster than that of any other cancers worldwide.15 Therefore we need to design a fluorescent analogous of the target compound 10. The p-nitrobenzenesulfonyl group was interchanged with the dansyl group as a chromophore because of its similarity (15, Fig. 2). Moreover, a fluorescence study of some anti-tumour drugs with a dansyl group in their structure that present no toxicity in vivo16 confirms the benefit of the use of this chromophore in the design. We also include the dansyl derivative with n = 1 (14) in order to complete the SAR studies. For the synthesis, see Scheme S1,† R1 = B, and R2 = C and D. We can establish the following SARs for all the target compounds 4–17: the IC50 values of compounds with n = 2 (8–10, 15, 17) are lower than those of compounds with n = 1 (4–6, 14–16) and this fact might be attributed to the increase of lipophilicity.17 The most active compounds against all cell lines assayed are the 6-chloro (8), 6-bromo (9) and 2,6-dichloropurine (10, 15–17) derivatives, as it has been previously reported.18 The N-3 adenine isomer (12) is slightly more active than the N-9 one (11). The 5-FU derivative (13) is the least active as anti-proliferative agent against all the cell lines. It should be pointed out that all the target compounds show better anti-proliferative activity against the human melanoma cell line with sub-micromolar IC50 values. Compound 10 is the best anti-proliferative agent of these p-nitrobenzenesulfonyl derivatives with an IC50 of 134 nM against the A-375 cell line. Dansyl derivatives (14–17) are also potent anti-proliferative agents against all cell lines with IC50 values between 0.3–3 μM, with the sole exception of 14 against the MCF-7 cell line (IC50 = 10.40 μM). Interestingly, when we tested the anti-proliferative activities for 8 and 10 on the A-375 isolated cell line, based on its ALDH activity, it was found that 10 shows a potent anti-proliferative activity against this isolated subpopulation (Table S2†).
The effects on cell cycle distribution of the most active compounds (4–6, 8–10, 14–17) were analyzed by flow cytometry for 24 and 48 h at 3 × IC50 in the A-375 cell line (Table S3†). Compounds 5 and 8–10 provoked a G2/M cell cycle arrest at the expense of the other two phases (26.60%, 34.40%, 37.27% and 21.36%, respectively, in contrast to the 8.71% of the control). The induction of the G2/M cell cycle arrest by three compounds previously synthesized by us was associated with increased phosphorylation of eIF2α in human breast cancer cells.19
We then analyzed the apoptotic effects of the most active compounds (4–6, 8–10, 14–17) in the A-375 cell line by flow cytometry after 24 and 48 h of treatment, in order to study the mechanisms of their anti-tumour activity. The results are shown in Table S4.† The most apoptotic compounds are 8–10, that are also the most active as anti-proliferative agents.
Microscopy is a useful tool to study drug distribution in biological tissues.20 We subsequently investigated the in vitro and in vivo distribution of 15, which is the fluorescent analogous of 10, in three tumour cell lines, in mice organs and tissues. MCF-7 breast cancer cells, HCT-116 colon cancer cells and A-375 melanoma cells were treated with compound 15 and absorbance was measured from 30 min to 2 h. Our results showed an increase in uptake quantities in treated cells that was time-dependent, being higher for MCF-7 at 30 min and 1 h. However, not significant differences were found after 2 h of treatment with 15 for the three tumour cell lines (Fig. 3A).
 | ||
| Fig. 3 (A) In vitro distribution of 15 in MCF-7 breast cancer cells, HCT-116 colon cancer cells and A-375 melanoma. (B) Confocal imaging in A-375 melanoma cells treated with a concentration of 10 μM of 15. | ||
Clearly, it is important to directly monitor the means by which 15 penetrates the cell membrane and whether it penetrates cell nuclei. To study the entry and distribution of 15, confocal imaging was used in the A-375 melanoma treated-cells (Fig. 3B, Movies S1 and S2†). Images show that after 1 h, compound 15 was internalized in a high percentage of cells and it was distributed in the perinuclear zone. After 2 h most of the cells showed enhanced fluorescence intensity and in some of them, 15 was located inside the nucleus. After 4 h a significant fraction of 15 was identified inside the cell nuclei with additional portions surrounding the perinuclear region (Fig. 4). Molecules observed in the cytoplasm appear as bright blue spots, suggesting that they were embedded by cells via endocytosis.
 | ||
| Fig. 4 Dansyl distribution inside the cell nuclei (arrows) of 15 after 4 h. | ||
Toxicity was determined after selecting the most active compound (10) that presents an IC50 value of 0.134 μM against A-375 cells. Its fluorescent analogous 15 was included in this study. We examined the acute-toxicity profile of 10 and 15 in BALB/c mice when they were administered in a single oral bolus each one (n = 40) at doses levels of 50–250 mg kg−1 for a week. Compounds 10 and 15 were non-toxic, even at the highest bolus dose of 250 mg kg−1 after 2 weeks. All the 40 10-treated and 15-treated mice remained healthy throughout the 14 day observation period with no evidence of morbidity.
For in vivo organ distribution analysis, mice were treated with 10 and 15 (100 mg kg−1) by oral administration and fluorescence was detected in the organs extracted by necropsy after 6 h (Fig. 5A).
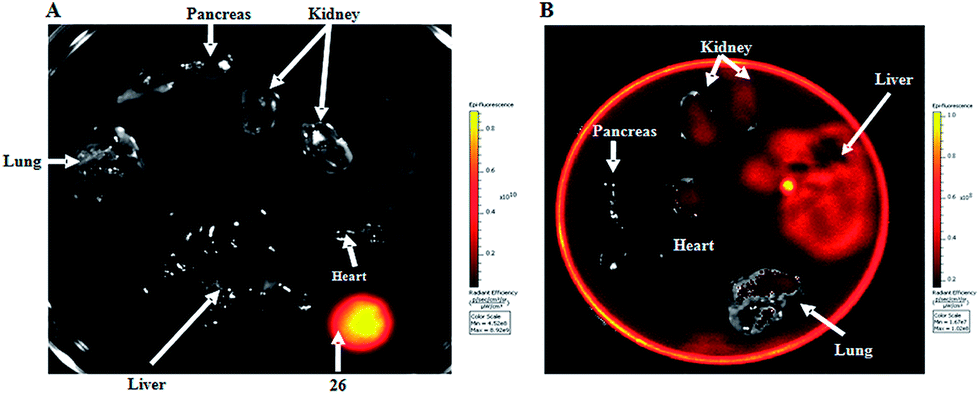 | ||
| Fig. 5 In vivo distribution study of 10 and 15 using the IVIS spectrum imaging system. (A) Distribution of 10 in several organs. A drop of 15 was used as positive control. (B) Distribution of 15 in several organs. | ||
Fluorescence intensity was high in all organs and tissues of mice treated with 15, with the exception of pancreas and lungs (Fig. 5B). Fluorescence intensity was not detected in mice treated with 10, used as a control (Fig. 5A). Results confirm that the drug is absorbed by the oral route of administration with systemic distribution. Furthermore the drug accumulates mainly in liver and kidney, the organs associated to drug metabolism and excretion, respectively.
Conclusions
A series of p-nitrobenzenesulfonyl and dansyl derivatives have been designed, synthesized and biologically evaluated as anti-tumour agents. Derivatives with a 6-bromo-, 6-chloro- and 2,6-dichloro-purine show better anti-proliferative activities against all the assayed cell lines than the adenine and 5-FU derivatives. Compounds with a dimethylene linker (n = 2) are better anti-proliferative agents than those with a methylene linker (n = 1). 2,6-Dichloropurine derivative with n = 2 and the p-nitrobenzenesulfonyl moiety (10) is the most active compound as an anti-proliferative agent with an IC50 of 134 nM against the A-375 cell line. In the A-375 ALDH+ CSC subpopulation 10 shows a high anti-proliferative activity. The most apoptotic compounds are the purine derivatives with n = 2 and they provoked a G2/M cell cycle arrest. Moreover, the 6-bromopurine derivative (9) is the most potent apoptosis inductor so far reported by us.The fluorescence of N-9 2,6-dichloropurine derivative with n = 2 (15) was detected by fluorimetric assays in cell cultures. Confocal imaging showed fluorescent N-9 dichloropurine derivative with n = 2 distribution in cell. The fluorescent compound seems to be incorporated to cell by an endocytic process. We observed vesicle transportation through cell cytoplasm and these vesicles accumulate in the perinuclear zone, and at late time (4 h) inside nuclei. It has been demonstrated that 10 and its fluorescent analogue 15 are nontoxic to BALB/c mice, even at the highest bolus dose of 250 mg kg−1 after 2 weeks. Compound 15 is absorbed by the oral route of administration with systemic distribution. Furthermore the drug accumulates mainly in liver and kidney, the organs associated to drug metabolism and excretion respectively. Nevertheless, deepest studies such as ADME assays are necessary to obtain a more accurate distribution of 15. Our findings support the potent anti-tumour ability of the fluorescent derivative (15) which has permitted to conduct single-cell imaging of this drug upon binding to its targets in differentiated bulk tumour cells and CSCs. These results encourage further studies in molecular CSC pathways and suggest that this molecule could be a new CSC-targeted drug.
Acknowledgements
We thank the Instituto de Salud Carlos III through projects no. PI10/00592 and PI10/02295 and the European Regional Development Fund for financial supports. The award of a grant from the Ministerio de Educación to F. M. is gratefully acknowledged.Notes and references
- V. Tirino, V. Desiderio, F. Paino, A. de Rosa, F. Papaccio, M. La Noce, L. Laino, F. de Francesco and G. Papaccio, FASEB J., 2013, 27, 13 CrossRef CAS PubMed.
- L. C. López-Cara, A. Conejo-García, J. A. Marchal, G. Macchione, O. Cruz-López, H. Boulaiz, M. A. García, F. Rodríguez-Serrano, A. Ramírez, C. Cativiela, A. I. Jiménez, J. M. García-Ruiz, D. Choquesillo-Lazarte, A. Aránega and J. M. Campos, Eur. J. Med. Chem., 2011, 46, 249 CrossRef PubMed.
- O. Caba, M. Díaz-Gavilán, F. Rodríguez-Serrano, H. Boulaiz, A. Aránega, M. A. Gallo, J. A. Marchal and J. M. Campos, Eur. J. Med. Chem., 2011, 46, 3802 CrossRef CAS PubMed.
- J. A. Marchal, E. Carrasco, A. Ramírez, G. Jiménez, C. Olmedo, M. Perán, A. Agil, A. Conejo-García, O. Cruz-López, J. M. Campos and M. A. García, Drug Des., Dev. Ther., 2013, 7, 1301 Search PubMed.
- A. Ramírez, H. Boulaiz, C. Morata-Tarifa, M. Perán, G. Jiménez, M. Picón-Ruiz, A. Agil, O. Cruz-López, A. Conejo-García, J. M. Campos, A. Sánchez, M. A. García and J. A. Marchal, Oncotarget, 2014, 5, 3590 CrossRef.
- M. Annette, S. Patil, N. S. Malik and K. D. Panghavane, PCT Int. Appl., WO 2012081039 A1 20120621, 2012.
- M. Díaz-Gavilán, F. Rodríguez-Serrano, J. A. Gómez-Vidal, J. A. Marchal, A. Aránega, M. A. Gallo, A. Espinosa and J. M. Campos, Tetrahedron, 2004, 60, 11547 CrossRef PubMed.
- K. Bowden and P. R. Williams, J. Chem. Soc., Perkin Trans. 2, 1991, 215 RSC.
- L. Fitzgerald, R. L. Blakeley and B. Zerner, Chem. Lett., 1984, 29 CrossRef CAS.
- A. Conejo-García, M. C. Núñez, J. A. Marchal, F. Rodríguez-Serrano, A. Aránega, M. A. Gallo, A. Espinosa and J. M. Campos, Eur. J. Med. Chem., 2008, 43, 1742 CrossRef PubMed.
- M. E. García-Rubiño, M. C. Núñez-Carretero, D. Choquesillo-Lazarte, J. M. García-Ruiz, Y. Madrid and J. M. Campos, RSC Adv., 2014, 4, 22425 RSC.
- N. S. Whitea and R. J. Erringtonb, Adv. Drug Delivery Rev., 2005, 57, 17 CrossRef PubMed.
- H. M. Coley, W. B. Amos and P. R. Tewntyman, Br. J. Cancer, 1993, 67, 1316 CrossRef CAS PubMed.
- M. P. Lillo, O. Cañadas and R. E. Dale, Biochemistry, 2002, 41, 12436 CrossRef CAS PubMed.
- M. S. Goldberg, J. T. Doucette, H. W. Lim, J. Spencer, J. A. Carucci and D. S. Rigel, J. Am. Acad. Dermatol., 2007, 57, 60 CrossRef PubMed.
- C. H. Chui, Q. Wang, W. C. Chow, M. C. Yuen, K. L. Wong, W. M. Kwok, G. Y. Cheng, R. S. Wong, S. W. Tong, K. W. Chan, F. Y. Lau, P. B. Lai, K. H. Lam, E. Fabbri, X. M. Tao, R. Gambari and W. Y. Wong, Chem. Commun., 2010, 46, 3538 RSC.
- R. Sánchez-Martín, J. M. Campos, A. Conejo-García, O. Cruz-López, M. Báñez-Coronel, A. Rodríguez-González, M. A. Gallo, J. C. Lacal and A. Espinosa, J. Med. Chem., 2005, 48, 3354 CrossRef PubMed.
- M. C. Núñez, F. Rodríguez-Serrano, J. A. Marchal, O. Caba, A. Aránega, M. A. Gallo, A. Espinosa and J. M. Campos, Tetrahedron, 2007, 63, 183 CrossRef PubMed.
- A. Conejo-García, M. E. García-Rubiño, J. A. Marchal, M. C. Núñez, A. Ramírez, S. Cimino, M. A. García, A. Aránega, M. A. Gallo and J. M. Campos, Eur. J. Med. Chem., 2011, 46, 3795 CrossRef PubMed.
- C. A. Thorling, Y. Dancik, C. W. Hupple, G. Medley, X. Liu, A. V. Zvyagin, T. A. Robertson, F. J. Burczynski and M. S. Roberts, J. Biomed. Opt., 2011, 16, 086013 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra13946e |
| This journal is © The Royal Society of Chemistry 2015 |