
Ultrasound mediated efficient synthesis of spironaphthoquinolines†
Abstract
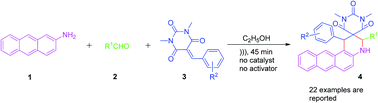
An atom-economical synthesis of spironaphthoquinolines from a mixture of 2-aminoanthracene, aldehyde and a Knoevenagel condensation product was developed. The association of the method with the use of ultrasound and its procedural simplicity makes it an attractive protocol for the formation of novel and important heterocycles.
 Please wait while we load your content...
Please wait while we load your content...

