DOI:
10.1039/C5RA08609D
(Paper)
RSC Adv., 2015,
5, 68829-68838
Protein–ligand interaction study of signal transducer smoothened protein with different drugs: molecular docking and QM/MM calculations†
Received
8th May 2015
, Accepted 30th July 2015
First published on 30th July 2015
Abstract
In this work, the interaction of signal transducer smoothened (SMO) protein, one of the main members of the Hedgehog (Hh) signaling pathway, with different ligands was studied. Seven hundred molecules, known as SMO ligands in literature, were selected for the docking screens. The site of the antitumor agent LY2940680, present in the X-ray crystal structure of the SMO protein, was selected as the binding site to study the interaction of the selected ligands with the protein. Docking screens only showed ten ligands that can be considered as favorable ligands based on their calculated free binding energies. The amino acid residues responsible for interacting with the ligands were identified. Quantum mechanics/molecular mechanics (QM/MM) calculations were also performed on the structure of the protein interacting with the favorable ligands to obtain more accurate results for their electronic interaction energies. The contribution of the van der Waals (vdW) and electrostatic interaction in the calculated electronic interaction energy of each ligand were calculated by QM/MM calculations. In addition, the effect of the polarization of the wave function of ligand in the active site of the protein on its electronic interaction with the protein was also studied.
1. Introduction
The Hh signaling pathway is a key regulator of cell proliferation and organ differentiation during embryonic development. Unusual Hh signaling in adults is associated with the initiation of several human cancers including basal-cell carcinoma (BCC), medulloblastoma, small-cell lung cancer (SCLC) and pancreatic a denocarcinoma as well as tumor growth and progression.1 Therefore, inhibition of the over-activated Hh pathway has become an interesting target for drug discovery.2 Main constituents of the Hh pathway are the Hh ligand, sonic Hedgehog (SHh), desert Hedgehog (DHh), Indian Hedgehog (IHh), the 12-pass trans membrane receptor Patched (Ptch), the 7-pass trans membrane receptor SMO, suppressor of fused (SUFU) and transcription factor Gli.3 In the absence of the Hh ligand, Ptch exerts an inhibitory effect on SMO and no downstream signaling occurs which means that SMO is in its inactive state while, when the Hh ligand binds to Ptch, SMO becomes activated. Activation of SMO is followed by its interaction with SUFU and subsequent translocation of Gli and then expression of Hh target genes which results in tumor cells proliferation.
Currently, SMO ligands are the best drug candidates which are applied as clinical trials for inhibiting the Hh pathway.4,5 Cyclopamine is the first known Hh inhibitor which directly binds to the heptahelical bundle of SMO protein. Cyclopamine also can reverse the retention of partially misfolded SMO in the endoplasmic reticulum, presumably through binding-mediated effects on protein conformation.6 There is one active binding site in the structure of SMO protein which is used for binding all of the SMO molecule antagonists and agonists to it.7 Although, it has been reported a large number of synthesized organic compounds as SMO ligands in literature but, the crystal structure of SMO was not available in literature until 2013. Wang et al.8 determined the crystal structure of the human SMO protein for the first time. Knowing the crystal structure of SMO is useful to perform molecular docking study on the different SMO ligands reported in literature to calculate their interaction with the protein and find their efficiency in the Hh signaling pathway.
Calculation of the ligand-receptor binding energy by molecular docking has provided a powerful tool for docking screens of the molecule libraries.9 In this work, the docking screens of more than 700 molecules, proposed in literature as SMO ligands, were examined by docking method to select favor ligands based on their calculated binding free energies. The ligands used in the docking screens were taken from Mahindroo et al.,2 Miller-Moslin et al.,10 Ishibashi et al.,11 Taber et al.,12 Ruat et al.,13 Lee et al.,14 Chang et al.,15 Sauvage et al.,16 Giannis et al.,17 Hipskind et al.,18 Dandawate et al.,19 Brunton et al.,20 Beachy et al.,21 Dai et al.22 and Austin et al.23 There is complete information about the medicinal chemistry and clinical aspects of the selected ligands in the ref. 2 and 10–23 which are not explained here more. Although, there are many papers on the medicinal chemistry and clinical aspects of the SMO inhibitors in literature but, there is no theoretical work on the interaction of inhibitors with the SMO protein in literature due to the lack of the X-ray structure of this protein. The docking screens were performed using AutoDock Vina software.24 QM/MM methodology, considering a realistic representation for the ligand, was also used for calculating the electronic interaction energies of the favorable ligands, selected by docking screens, with SMO protein. In QM/MM calculations, the ligand and SMO protein was considered as quantum mechanical and molecular mechanical part of system, respectively.
2. Methods
2.1. Structural preparation of the SMO protein and ligands
The crystal structure of the target protein, SMO receptor (PDB ID 4JKV) was taken from the Protein Data Bank.25 The water molecules in the protein crystal structure were removed using the AutoDock Tools 1.5.4 software.26 There were no water molecules near the antitumor agent LY2940680 in the X-ray structure of the SMO protein. Also, there were missing hydrogen atoms in the structure of amino acid residues including Lys15, Lys539, Arg547, Arg291, Lys356, Arg546 and Arg257 of chain A and Arg551, Lys344, Arg547, Lys356, Lys539, Arg257, Arg261, Asn258 and Lys444 of chain B of SMO protein. To complete the structure of SMO protein for the docking calculations, the missing hydrogen atoms in the structure of protein were added. Kollman united atom charges were assigned and atomic solvation parameters were added. Before performing docking, the structures of ligands were optimized using MM2 method implemented in ChemOffice package.27 For polar hydrogen atoms, the Gasteiger type partial charges were assigned and nonpolar hydrogen atoms were merged and the number of torsions was set.
2.2. Binding site identification
The binding site of antitumor agent LY2940680 in the X-ray structure of the SMO protein, was considered for the docking of the considered ligands (see Fig. 1). There are two similar binding sites in two chains (A and B) of this protein but, all of the calculations in this work were performed only on the binding site of chain B of SMO. It should be mentioned that the SMO protein is crystallized as parallel dimer in the crystallographic asymmetric unit. From Fig. 1 of ref. 8 is obvious that there is no difference between the binding site on chains A and B of SMO protein. Before performing docking calculations, it was decided to analyze the binding site of antitumor agent LY2940680 using LigPlot + software.28 The calculations revealed that LY2940680 interacts with amino acid residues including Met301, Leu221, Asn219, Asp384, Pro513, Tyr394, Glu518, Ser384, Val386, Ser387, Met230, Arg400, Phe391, Asn521, Trp281 and Leu522 (see Fig. 2). As seen, there are two hydrogen bonds between the ligand and Asn219, Arg400 residues with bond lengths of 2.81 and 3.17 Å, respectively. The same binding site was also found for this receptor using the Q-SiteFinder method29 with the volume of 798 Å3. Q-SiteFinder also predicted the ligand binding site by binding hydrophobic probes to the protein and finding clusters of probes with the most favorable binding energy. The shape and location of this binding site has been shown in Fig. 1. This result was also confirmed by carrying out blind docking on the protein using Auto Dock Vina.24
 |
| | Fig. 1 The location and shape of the ligand binding site of LY2940680 on chain B of SMO protein determined using Q-SiteFinder method.29 | |
 |
| | Fig. 2 Schematic representation of hydrogen bondings and vdW interaction of LY2940680 ligand with SMO protein. | |
2.3. Docking screens
The docking screens of 716 ligands, proposed in literature as SMO ligands (see Table S1; ESI†), were performed with AutoDock Vina and PyRx virtual screening tool.30 A search space size of the 26 × 20 × 26 Å and an exhaustiveness of 8 and maximum number of binding modes of 9 were defined for the program. The geometries of ligands and the structure of protein were considered flexible and rigid, respectively. The calculated binding free energies (ΔGb) of the ligands were in the range of −14.8 to 20.1 kcal mol−1. Some of the ligands had the positive binding free energies and could not place in the active site of protein although, they have been proposed as the SMO ligands in literature. On the other hand, most of the proposed ligands had small negative free binding energies. Ten ligands with the high binding free energies were selected for the QM/MM calculations because this kind of calculation is so time consuming, especially, when the receptor is protein with many atoms (see Fig. 3). For further confirmation, the docking screens was also performed using Autodock 4.2 (ref. 26) and ArgusLab 4.0.1 (ref. 31) softwares, separately and the same ligands as those predicted by AutoDock Vina were predicted. In docking with AutoDock 4.2, the Lamarckian genetic algorithm32 was used. A grid box with XYZ dimensions of 104 Å × 68 Å × 90 Å, respectively with grid point spacing of 0.375 Å was generated. Within this grid box, auto grid computed the affinity maps of the present atoms as well as electrostatic map. The protein structure was considered as rigid and the ligands were considered to be flexible. Every docking result was derived from 10 genetic algorithm runs which terminated after a maximum of 2.5 × 106 energy evaluations in a maximum of 2.7 × 104 generations with population size of 150 individuals and a rate of gene mutation of 0.02 and crossover rate of 0.8. The results were evaluated by ranking various complexes towards the predicted binding energy. The cluster analysis was subsequently accomplished on the basis of root mean square deviation values with respect to the starting geometry. The docked conformation with the most favorable binding free energy and more populated cluster was selected as the best result. In docking using ArgusLab software, the AScore function which is an empirical scoring function was used for the docking calculations. The binding site box dimensions in x, y and z directions were 40 × 32.5 × 35.3 Å3, respectively and a grid resolution of 0.4 Å was used. The ligands were considered as flexible and they were docked to the rigid protein with ArgusDock docking engine. It should be mentioned that the Autodock 4.2 and ArgusLab 4.0.1 softwares proposed the selected ligands same as those predicted by AutoDock Vina. In addition, the interaction of the antitumor agent LY2940680 in the X-ray structure of the SMO protein was also calculated by AutoDock Vina, Autodock 4.2 and ArgusLab 4.0.1, separately. In addition, BINANA algorithm33 was employed to analyze the binding free energies of ligands obtained from the docking calculations. This algorithm can provides information about the types and numbers of interactions which contribute to the ligand binding.
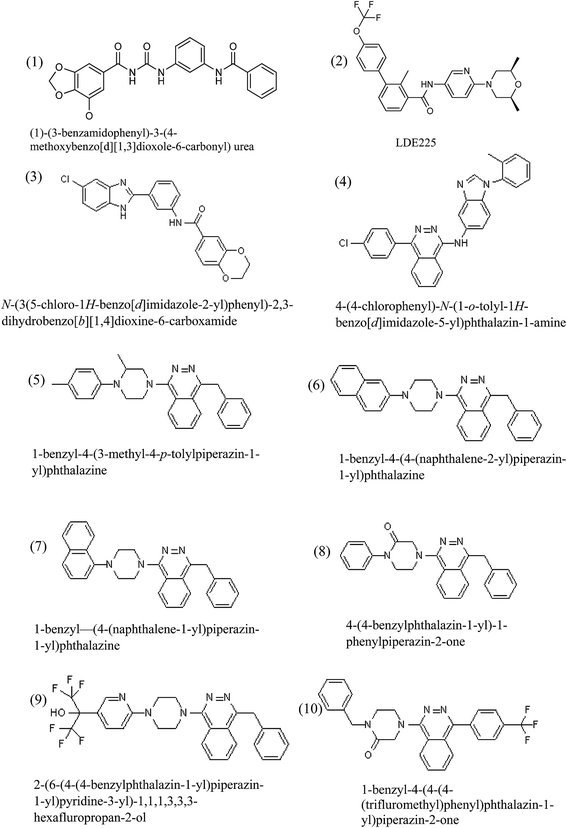 |
| | Fig. 3 The molecular structures of ten ligands, selected after docking screens, with favorable interactions with SMO protein and used for QM/MM studies in this work. | |
2.4. QM/MM approach
Our own N-layered Integrated molecular Orbital and molecular Mechanics (ONIOM) methodology34 was employed to perform QM/MM calculations and obtain the electronic interaction energies of the selected ligands with SMO protein. The ONIOM calculations were carried out using Gaussian quantum chemistry package (Gaussian 09, revision A.01).35 ONIOM scheme is more general in the sense that it can combine any number of molecular orbital methods as well as molecular mechanics methods.36–43 This method enables different ab initio or semi-empirical methods along with molecular mechanic method to be applied to different parts of a molecule or system such as biomolecules to produce reliable geometry and energy at the reduced computational time. For example, the study of the interaction of different molecules, drugs and ligands with DNA, membranes and proteins are the suitable case which can be performed by this method.
The total electronic energy of ligand and protein (Etotal) in ONIOM calculations is obtained from the following equation (subtractive QM/MM scheme):34
| | |
Etotal = EQMmodel + EMMreal − EMMmodel
| (1) |
where
EMMreal is the MM energy of the entire system (protein + ligand), called real system;
EQMmodel is the QM energy of a part of real system that has main chemical interest, called model part (ligand); and
EMMmodel is the MM energy of the model part (ligand).
Etotal can also be decomposed through the following equation (additive QM/MM scheme):
| | |
Etotal = EQMhigh-layer + EMMlow-layer + EQM/MMint
| (2) |
where
EQMhigh-layer is the energy of the quantum part of system (ligand),
EMMlow-layer is the energy of low layer part of system (protein) and
EQM/MMint is the interaction energy between the high layer (ligand) and low layer (protein) of system. It should be noted that
Etotal has been calculated using subtractive QM/MM scheme in this work.
The interaction energy obtained based on the QM/MM calculations (EQM/MMint) can be divided to three terms including ΔEvdw, ΔEelec and ΔEpol.44 ΔEvdw is the contribution of vdW interaction between ligand and protein, ΔEelec is the contribution of electrostatic interaction of unpolarized ligand at its gas phase charge distribution which also accounts hydrogen bonding and ΔEpol is a part of interaction which is related to the polarization of the wave function of ligand in the active site of protein. In fact, the wave function of ligand is polarized by the electrostatic charge distribution of MM region. For this purpose, the atomic charges of MM region were implemented in the QM Hamiltonian of ligand to consider the effect of the electrostatic field of MM region on the wave function of ligand. It is notable that the last term (ΔEpol) of interaction is absent in the docking calculations.
It should be mentioned that the interaction energy between a protein and ligand is calculated in the docking softwares using molecular mechanic method considering an appropriate force field. This means that both protein and ligand are considered molecular mechanically. In the ONIOM method, it is possible to consider the ligand as quantum mechanical part of the system and have a more realistic description of the protein along with ligand. The other aim for using ONIOM method in this work is to evaluate the site of SMO protein for binding to the selected ligands based on the calculated electronic interaction energies and not free energy binding energy. In the other words, the final structures of ligand + SMO, obtained from the docking calculations, are evaluated based on their calculated electronic interaction energy using QM/MM method.
3. Results and discussion
3.1. Docking results and analysis
Table 1 lists the calculated binding free energies of the selected ligands, shown in Fig. 3, to SMO protein on specified binding site calculated by three different softwares including AutoDock Vina, AutoDock 4.2 and ArgusLab. It is seen that AutoDock Vina software predicts higher binding free energy for ten ligands compared to the other softwares. In addition, the calculated binding energy of antitumor agent LY2940680 in the X-ray structure of the SMO was also calculated and included in this table. It is seen that the calculated binding free energies of the selected ligands are comparable with the binding free energy of LY2940680. The reason for the difference among the calculated binding free energy of a ligand with different softwares is related to the difference in the docking algorithms and scoring functions used by these softwares. The calculated binding free energies obtained using the Auto dock Vina could be more reasonable and accurate compared to those obtained using the other two docking softwares. Because, Vina uses a sophisticated gradient optimization method in its local optimization procedure and the calculation of the gradient effectively gives the optimization algorithm a “sense of direction” from a single evaluation.24 The evaluation of the accuracy of Vina during flexible redocking of the 190 receptor–ligand complexes has been shown that the accuracy of the binding mode prediction of this software has been significantly improved compared to AutoDock 4 (ref. 24) when compared with experiment. The order of the interaction of ligands, shown in Fig. 3, based on their calculated binding free energies by AutoDock Vina, AutoDock 4.2 and ArgusLab are 4 > 6 > 2 > 1> 7 > 5> 8 > 3> 10 > 9, 6 > 7 >5 > 8 >2 > 1> 9 > 4 > 3 > 10 and 8 > 3 > 4 > 10 > 5 > 2 > 7 > 6 > 9 > 1, respectively.
Table 1 The binding free energies (ΔGb) of the ligands, selected by virtual screening (see Fig. 3), to the SMO protein on specified binding site calculated by three different software including AutoDock Vina, AutoDock 4.2 and ArgusLab. The numbering of ligands is according to Fig. 3
| Ligand no. |
ΔGb/kcal mol−1 |
| Auto dock Vina |
Argus lab |
Auto dock 4.2 |
| 1 |
−13.5 |
−9.12 |
−11.36 |
| 2 |
−13.6 |
−12.16 |
−11.86 |
| 3 |
−13.1 |
−13.16 |
−10.62 |
| 4 |
−14.8 |
−12.87 |
−10.95 |
| 5 |
−13.4 |
−12.63 |
−12.69 |
| 6 |
−14.3 |
−11.54 |
−13.66 |
| 7 |
−13.4 |
−11.92 |
−12.82 |
| 8 |
−13.3 |
−13.40 |
−12.36 |
| 9 |
−12.9 |
−10.31 |
−11.34 |
| 10 |
−13.1 |
−12.66 |
−10.17 |
| LY2940680 |
−14.4 |
−11.40 |
−12.09 |
The key interacting amino acid residues with the SMO ligands, obtained using pymol software, have been demonstrated in Fig. 4 and 5. It is observed that all ligands interact with Trp281, Asp384, Val386, Ser387, Tyr394 and Glu518 residues. Some of the residues are found to have interaction with most of the ligands, such as Met230 which has interaction with all selected ligands except for ligand no. 3 (see Fig. 3). Moreover, Asn219 residue is found to have interaction with ligands no. 2 to 9. On the other hand, some residues such as Leu515 have only interaction with ligand no. 2. Lys395 and Asp473 residues have only interactions with ligands no. 8 and 9.
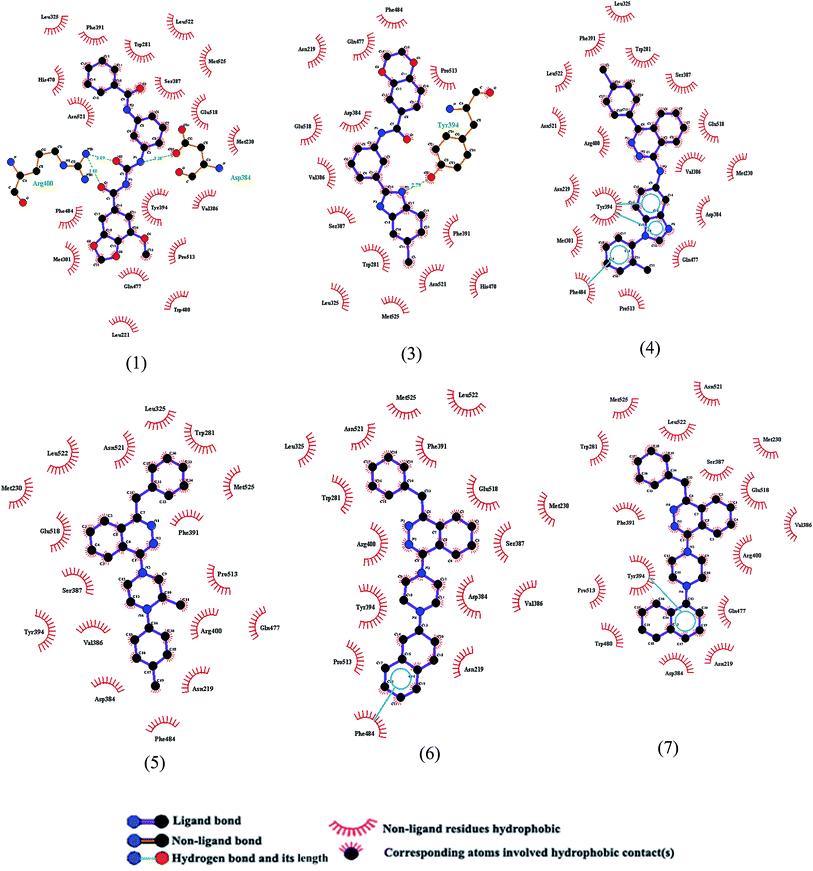 |
| | Fig. 4 Schematic representation of hydrogen bondings, vdW interactions and aromatic ring interaction of six selected ligands, shown in Fig. 3, with SMO protein obtained using pymol software. The labeling of ligands is based on Fig. 3. | |
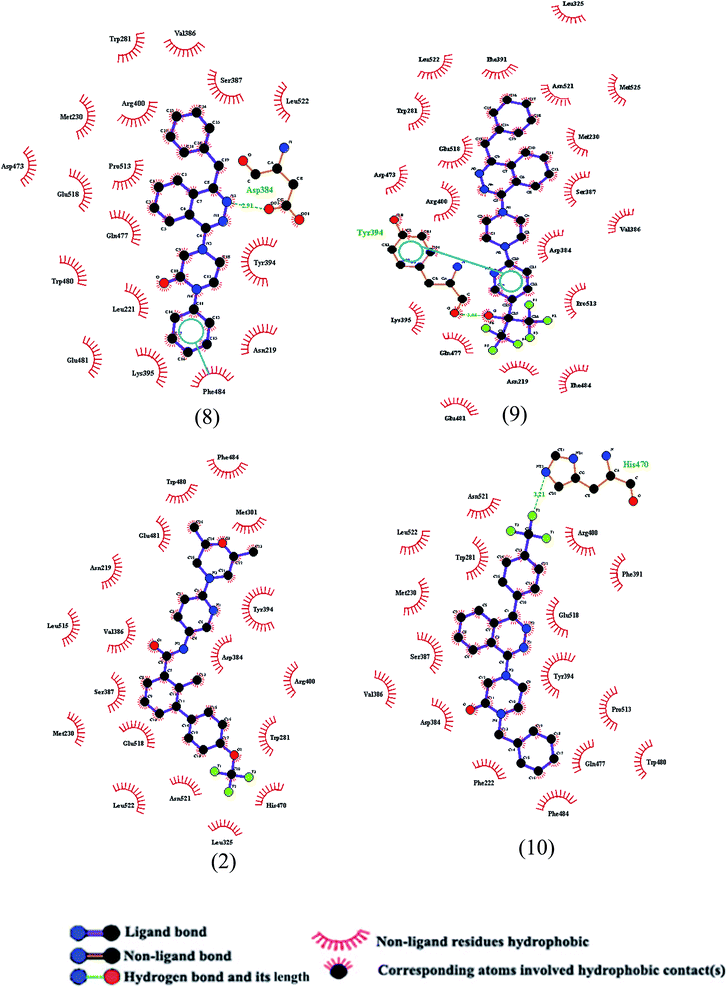 |
| | Fig. 5 Schematic representation of hydrogen bondings, vdW interactions and aromatic ring interaction of four selected ligands, shown in Fig. 3, with SMO protein obtained using pymol software. The labeling of ligands is based on Fig. 3. | |
In order to analyze the binding free energies of ligands obtained from docking calculations, BINANA algorithm33 was used. This algorithm provides information about the types and numbers of interactions which contribute to ligand binding. This program identifies key binding characteristics like hydrogen bonds and π interactions. The distance cutoff for the interactions of the type of π–π, cation-π, hydrophobic, T-stacking and hydrogen bond are 7.5, 6.0, 4.0, 5.0 and 4.0 angstroms, respectively.33 These distances cutoff are defined as default in the BINANA algorithm. Table 2 summarizes the type and number of interactions between the selected SMO ligands and the amino acid residues. The most favorable binding energy calculated by AutoDock Vina is related to ligand no. 4 which could be attributed to the greatest number of π–π interactions (see Table 2). There are three π–π interactions between this ligand and amino acid residues within 7.5 Å. These interactions have been visualized in Fig. 6 by VMD 1.8.7 software.45 These π–π interactions are between phenyl ring of the Phe484 amino acid (the upper ring in Fig. 6) and tolyl ring of the ligand, and also between phenyl ring of Tyr394 amino acid (the lower ring in Fig. 6) and benzimidazol part of the ligand.
Table 2 Type and number of interactions, calculated by BINANA algorithm, for the selected ligands reported in Table 1. The numbering of ligands is according to Fig. 3
| Ligand no. |
Number of interactions |
| H-bond |
Hydrophobic contact |
π–π stacking |
T-stacking |
Cation-π |
| 1 |
2 |
46 |
0 |
3 |
1 |
| 2 |
1 |
52 |
0 |
2 |
1 |
| 3 |
2 |
40 |
0 |
2 |
2 |
| 4 |
2 |
58 |
3 |
3 |
1 |
| 5 |
3 |
60 |
0 |
3 |
1 |
| 6 |
3 |
55 |
1 |
3 |
1 |
| 7 |
3 |
58 |
1 |
3 |
1 |
| 8 |
0 |
53 |
1 |
0 |
0 |
| 9 |
3 |
62 |
1 |
3 |
1 |
| 10 |
3 |
52 |
0 |
1 |
1 |
 |
| | Fig. 6 π–π interactions between ligand no. 4 (see Table 2) and surrounding amino acid residues. | |
3.2. QM/MM results
The structures of the selected ligands (shown in Fig. 3) + SMO protein, obtained from the docking calculation by AutoDock Vina, were used as initial structures for ONIOM calculations. A two layers ONIOM method was selected for QM/MM calculations in this work so that the protein was considered as low layer and the ligand as high layer. MM method was selected for the low layer and the density functional theory (DFT) method employing B3LYP functional and 6-31+G(d) basis set was used for the high layer. Fig. 7 shows the ONIOM layer assignment of SMO protein and a typical ligand taken from Fig. 3. The universal force field (UFF) was used for the MM region. Therefore, two-layer ONIOM calculations (B3LYP/6-31+G(d):UFF) was performed. To obtain more accurate values for the electronic interaction energies, the ligands were considered flexible in the active site of protein in the QM/MM calculations. The optimization of the ligands in the active site of protein, using QM/MM calculations, were performed in three different conditions including (i) considering only vdW interaction between ligand and protein (ii) considering both vdW and electrostatic interaction between ligand and protein (iii) considering the vdW and electrostatic interaction between the ligand and protein plus electronic embedding which incorporates the partial charges of the MM region into the Hamiltonian of ligand. Electronic embedding causes that the polarization of the wave function of ligand in the presence of the electrostatic field of protein is considered.
 |
| | Fig. 7 ONIOM layer definition for modeling SMO protein and ligand. | |
Table 3 reports the electronic interaction energies of the ligands with SMO protein obtained from the ONIOM calculations at the (B3LYP/6-31+G(d):UFF) level of theory. The first column of Table 3 shows the vdW interaction between the ligands and protein. It is seen that the vdW interaction for all of the ligands is attractive. The vdW interaction energies (ΔEvdw) of ligands no. 2, 4, 6 and 7 (−65.13, −63.48,−65.27 and −65.23 kcal mol−1, respectively) are more close to the value of ΔEvdw of LY2940680 (−68.34 kcal mol−1) compared to the other ligands. The second column of Table 3 reports the sum of ΔEvdw and the electrostatic interaction energy (ΔEelec) for the ligands. The numbers in the parenthesis of this column show the values of ΔEelec for the ligands. It can be seen that ΔEelec is attractive for all of the ligands and ligands no. 2, 9 and 10 have higher ΔEelec compared to the other ligands. The ligand no. 9 has the highest electrostatic attraction with the protein which is also higher than that of LY2940680. It is interesting to notice that the ligands containing F atom in their structure have considerable ΔEelec with protein. Ligand no. 9 has the highest value of (ΔEelec + ΔEvdw) among the ligands shown in Fig. 3. The third column of Table 3 reports the sum of ΔEvdw, ΔEelec and the polarization interaction energy (ΔEpol) for the ligands and the values in parenthesis are ΔEpol. It can be seen that the value of ΔEpol is positive for all of the ligands except for ligand no. 6 (−0.69 kcal mol−1). The positive value of ΔEpol means that the electrostic field of protein polarizes the wave function of the ligand so that produces the repulsive interaction between the ligand and protein. Comparison of the second column with the third column of Table 3 show that the calculated value of ΔEpol of each ligand is comparable with its ΔEelec and higher value of ΔEelec is accompanied with the higher value of ΔEpol except for ligand no. 6 which its ΔEpol is attractive. For some ligands in Table 3, the absolute value of ΔEpol is greater than the absolute value of ΔEelec so that the repulsion due to the polarization completely cancel out the contribution of the attraction due to the electrostatic interaction between ligand and protein in the total electronic interaction energy (ΔEvdw + ΔEelec + ΔEpol). The total interaction energy of all ligands in Table 3 with SMO is negative (attractive) and ligand no. 6 has the best value of the electronic interaction energy with SMO protein (−74.94 kcal mol−1).
Table 3 The electronic interaction of the selected ligands (shown in Fig. 3) with SMO protein obtained from QM/MM calculations at the ONIOM (B3LYP/6-31+G(d):UFF) level of theory. The numbers in parenthesis in the second and third column show the ΔEelec and ΔEpol, respectively. The numbering of ligands is according to Fig. 3
| Ligand no. |
ΔEinta/kcal mol−1 |
ΔEintb/kcal mol−1 |
ΔEintc/kcal mol−1 |
| ΔEvdW. ΔEvdW + ΔEelec. ΔEvdW + ΔEelec + ΔEpol. ΔEelec. ΔEpol. |
| 1 |
−56.55 |
−66.29 (−9.74)d |
−57.46 (8.83)e |
| 2 |
−65.13 |
−109.76 (−44.63) |
−56.52 (53.24) |
| 3 |
−51.59 |
−60.95 (−9.36) |
−51.84 (9.11) |
| 4 |
−63.48 |
−73.78 (−10.30) |
−56.23 (17.55) |
| 5 |
−51.40 |
−74.13 (−22.73) |
−65.16 (8.97) |
| 6 |
−65.27 |
−74.25 (−8.98) |
−74.94 (−0.69) |
| 7 |
−65.23 |
−74.33 (−9.10) |
−65.84 (8.49) |
| 8 |
−51.73 |
−70.80 (−19.07) |
−55.89 (14.91) |
| 9 |
−59.97 |
−187.33 (−127.36) |
−58.98 (128.35) |
| 10 |
−55.19 |
−95.99 (−40.80) |
−52.98 (43.01) |
| LY2940680 |
−68.34 |
−129.08 (−60.74) |
−60.38 (68.70) |
4. Conclusion
The interactions of the different lignads (more than 700 molecules), known as SMO ligands in literature, were estimated by molecular docking calculations in this work. The docking screens were performed on over 700 ligands using three different docking softwares (Auto Dock Vina, Autodock 4.2 and ArgusLab 4.0.1), separately. The calculations showed that Trp281, Asp384, Val386, Ser387, Tyr394 and Glu518 residues in SMO protein have interaction with all considered ligands. QM/MM calculations, at ONIOM(B3LYP/6-31+G(d):UFF) level of theory, were employed to calculate the electronic interaction energies of ligands with the protein and evaluate the binding site of SMO protein for the selected ligands only based on their calculated electronic interaction energies. The decomposition of the electronic interaction energy to vdW and electrostatic interactions were performed for each ligand. It was found that the polarization of the wave functions of ligands by the electrostatic field of protein show itself as repulsive interaction in the electronic interaction and its value is comparable with ΔEelec except for ligand no. 6 in Table 3.
Acknowledgements
The authors thank Isfahan University of Technology for its financial support.
References
- M. Hebrok and M. P. Di Magliano, Nat. Rev. Cancer, 2003, 3, 903–911 CrossRef PubMed.
- N. Mahindroo, C. Punchihewa and N. Fujii, J. Med. Chem., 2009, 52, 3829–3854 CrossRef CAS PubMed.
- J. Taipale and M. Varjosalo, J. Cell Sci., 2007, 120, 3–6 Search PubMed.
- T. L. Lin and W. Matsui, Journal of OncoTargets and Therapy, 2012, 5, 47–58 CrossRef CAS PubMed.
- M. F. Strand, S. R. Wilson, J. L. Dembinski, D. D. Holsworth, A. Khvat, I. Okun, D. Petersen and S. Krauss, PLoS One, 2011, 6, e19904 CAS.
- P. A. Beachy, J. K. Chen, J. Taipale and M. K. Cooper, Genes Dev., 2001, 16, 2743–2748 Search PubMed.
- R. Rohatgi, D. F. Covey, S. Nachtergaele, L. K. Mydock, K. Krishnan, J. Rammohan and P. H. Schlesinger, Nat. Chem. Biol., 2012, 8, 211–220 CrossRef PubMed.
- C. Wang, H. Wu, V. Katritch, G. W. Han, X.-P. Huang, W. Liu, F. Y. Siu, B. L. Roth, V. Cherezov and R. C. Stevens, Nature, 2013, 497, 338–343 CrossRef CAS PubMed.
- T. Hou, Y. Li and S. Zhou, J. Chem. Inf. Model., 2013, 53, 982–996 CrossRef PubMed.
- K. Miller-Moslin, S. Peukert, R. K. Jain, M. A. McEwan, R. Karki, L. Llamas, N. Yusuff, F. He, Y. Li, Y. Sun, M. Dai, L. Perez, W. Michael, T. Sheng, H. Lei, R. Zhang, J. Williams, A. Bourret, A. Ramamurthy, J. Yuan, R. Guo, M. Matsumoto, A. Vattay, W. Maniara, A. Amaral, M. Dorsch and J. F. Kelleher, J. Med. Chem., 2009, 52, 3954 CrossRef CAS PubMed.
- M. Ishibashi, A. Shintani, K. Toume, Y. Rifai and M. A. Arai, J. Nat. Prod., 2010, 73, 1711–1713 CrossRef PubMed.
- D. F. Taber and P. W. DeMatteo, J. Org. Chem., 2012, 77, 4235–4241 CrossRef CAS PubMed.
- M. Ruat, M. Taddei, F. Manetti, A. Mann, A. Schoenfelder, E. Traiffort, H. Roudaut, H. Faure and A. Solinas, J. Med. Chem., 2012, 55, 1559–1571 CrossRef PubMed.
- X. Yang, Q. Shi, C.-Y. Lai, C.-Y. Chen, E. Ohkoshi, S.-C. Yang, C.-Y. Wang, K. F. Bastow, T.-S. Wu, S.-L. Pan, C.-M. Teng, P.-C. Yang and K.-H. Lee, J. Med. Chem., 2012, 55, 6751 CrossRef CAS PubMed.
- D. P. Walsh and Y.-T. Chang, Chem. Rev., 2006, 106(6), 2476–2530 CrossRef CAS PubMed.
- J. A. Low and F. J. de Sauvage, J. Clin. Oncol., 2010, 28, 5321–5326 CrossRef CAS PubMed.
- P. Heretsch, L. Tzagkaroulaki and A. Giannis, Angew. Chem., Int. Ed., 2010, 49, 3418–3427 CrossRef CAS PubMed.
- P. A. Hipskind, B. K. Patel and T. Wilson, WO2010147917A1, 2010.
- P. Dandawate, E. Khan, S. Padhye, H. Gaba, S. Sinha, J. Deshpande, K. V. Swamy, M. Khetmalas, A. Ahmad and F. H. Sarkar, Bioorg. Med. Chem. Lett., 2012, 22, 3104–3108 CrossRef CAS PubMed.
- S. A. Brunton, J. H. A. Stibbard, L. L. Rubin, L. I. Kruse, O. M. Guicherit, E. A. Boyd and S. Price, J. Med. Chem., 2008, 51, 1108–1110 CrossRef CAS PubMed.
- P. A. Beachy, J. K. Chen and A. J. Taipale, WO 2005/033288 A2, 2005.
- M. Dai, H. Feng, J. K. Rishi, R. Karki, J. Kelleher, J. Lei, L. Liamas, M. A. Mcewan, K. Miller-Moslin, L. B. Perez, S. Peukert and N. Yusuff, WO 2008/110611 A1, 2008.
- R. J. Austin, J. Kaizerman, B. Lucas, D. L. Mcminn and J. Powers, WO 2009/002469 A1, 2009.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
- http://www.rcsb.org/pdb/.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- http://www.cambridgesoft.com/Ensemble_for_Chemistry/ChemOffice/.
- A. C. Wallacel, R. A. Laskowski and J. M. Tornton, Protein Eng., 1995, 8, 127–134 CrossRef PubMed.
- A. Laurie and R. Jackson, Bioinformatics, 2005, 21, 1908–1916 CrossRef CAS PubMed.
- L. Wolf, PyRx. C&EN, 2009, 87, 31.
- M. A. Thompson, Molecular docking using ArgusLab, an efficient shape-based search algorithm and the AScore scoring function, in ACS meeting, Philadelphia 172, CINF 42, PA, 2004 Search PubMed.
- G. M. Morris, D. S. Goodsell, R. S. Halliday, R. Huey, W. E. Hart, R. K. Belew and A. J. Olson, J. Comput. Chem., 1998, 19, 1639–1662 CrossRef CAS.
- J. Durrant and J. McCammon, J. Mol. Graphics Modell., 2011, 29, 888–893 CrossRef CAS PubMed.
- S. Dapprich, I. Komáromi, K. S. Byun, K. Morokuma and M. J. Frisch, J. Mol. Struct.: THEOCHEM, 1999, 461–462, 1–21 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, revision A.01, Gaussian, Inc, Wallingford, 2009 Search PubMed.
- A. Altun, S. Yokoyama and K. Morokuma, J. Phys. Chem. B, 2008, 112, 6814–6827 CrossRef CAS PubMed.
- H. P. Hratchian, P. V. Parandekar, K. Raghavachari, M. J. Frisch and T. Vreven, J. Chem. Phys., 2008, 128, 034107 CrossRef PubMed.
- M. Svensson, S. R. Humbel, D. Froese, J. Matsubara, S. Sieber and K. J. Morokuma, J. Phys. Chem., 1996, 100, 19357–19363 CrossRef CAS.
- T. Vreven, B. Mennucci, C. O. Da Silva, K. Morokuma and J. Tomasi, J. Chem. Phys., 2001, 115, 62–72 CrossRef CAS PubMed.
- P. B. Karadakov and K. Morokuma, Chem. Phys. Lett., 2000, 317, 589–596 CrossRef CAS.
- N. Rega, S. S. Iyengar, G. A. Voth, H. B. Schlegel, T. Vreven and M. J. Frisch, J. Phys. Chem. B, 2004, 108, 4210–4220 CrossRef CAS.
- S. Humbel, S. Sieber and K. Morokuma, J. Chem. Phys., 1996, 105, 1959–1967 CrossRef CAS PubMed.
- T. Vreven and K. Morokuma, J. Comput. Chem., 2000, 21, 1419–1432 CrossRef CAS.
- T. Vreven, K. S. Byun, I. Komaromi, S. Dapprich, J. A. Montgomery Jr., K. Morokuma and M. J. Frisch, J. Chem. Theory Comput., 2006, 2, 815–826 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra08609d |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 






